

Negli ultimi anni il progresso in campo farmaceutico ha effettuato notevoli passi avanti, portando all’introduzione di farmaci sempre più innovativi e all’avvento dei farmaci biologici e della medicina personalizzata. La velocità con la quale vengono studiate e sviluppate le nuove alternative terapeutiche viene, però, frenata da procedure approvative lunghe e complesse che ritardano l’accesso dei pazienti ai nuovi trattamenti, talvolta salvavita.
Secondo un recente rapporto IQVIA pubblicato da EFPIA (European Federation of Pharmaceutical Industries and Associations), infatti, un nuovo farmaco è disponibile per i cittadini europei dopo una media di 426 giorni dall’approvazione in EMA (European Medicines Agency). Lo studio ha preso in esame 121 prodotti approvati con procedura centralizzata tra gennaio 2015 e dicembre 2017 e ha valutato il tasso di disponibilità (inteso come il numero di farmaci disponibili ai pazienti europei alla data del 19 dicembre 2018) e ritardo di accesso al mercato (inteso come il numero di giorni trascorsi dall’autorizzazione europea al termine delle procedure di autorizzazione nazionali) in 30 paesi europei.
In Europa il tempo medio di accesso al mercato di un nuovo farmaco è 426 giorni dall’approvazione in EMA
In termini di disponibilità dei farmaci l’Italia si colloca al quinto posto con il 79% dei 121 prodotti approvati in EMA disponibili alla data di cut-off dell’analisi. In testa alla classifica si trovano Regno Unito (88%), Germania (86%) e Austria (84%), mentre nelle ultime posizioni ci sono Macedonia (24%), Serbia (36%) e Lettonia (23%).
Per quanto riguarda i tempi di accesso al mercato l’Italia risulta meno virtuosa collocandosi al 14esimo posto, con una media di 402 giorni. Al primo posto si trova la Germania (119 giorni), seguita da Danimarca (146 giorni) e Svizzera (171 giorni), mentre in fondo alla classifica ci sono Serbia (925 giorni) e Lituania (726 giorni). La sottoanalisi, che ha preso in esame solo i farmaci oncologici, colloca l’Italia al 7° posto in termini di disponibilità con l’81% dei farmaci a disposizione e una durata media del processo di approvazione nazionale di 368 giorni (12° posto). Per quanto riguarda i farmaci orfani, invece, il tasso di disponibilità per l’Italia è del 76% (5° posto) mentre il tempo medio di approvazione è di 428 giorni (12° posto).
In Europa, a livello centralizzato e locale, sono numerose le iniziative intraprese negli ultimi anni per accelerare i tempi di accesso al mercato di farmaci che rispondono a importanti unmet needs, cercando di superare le incertezze in termini di efficacia e sicurezza derivanti dall’approvazione di molecole in fasi precoci di sviluppo.
Iter di approvazione di un nuovo farmaco
In Europa
Il Regolamento CE n. 726/2004, per l’istituzione di procedure comunitarie per l’autorizzazione e la sorveglianza dei medicinali per uso umano e veterinario, ha stabilito la necessità di rendere obbligatoria la procedura centralizzata di autorizzazione all’immissione in commercio (AIC), e la disponibilità immediata dopo l’approvazione in tutti gli stati membri, per tutti i medicinali:
- ad alta tecnologia, in particolare quelli derivati da biotecnologie;
- per terapia avanzata, quali la terapia genica, le terapie cellulari associate e la terapia somatica xenogenica;
- orfani;
- contenenti nuove sostanze attive aventi come indicazione terapeutica il trattamento della sindrome da immunodeficienza acquisita, del cancro, di disordini neurodegenerativi, del diabete, delle malattie autoimmuni e delle malattie virali.
È inoltre previsto l’accesso facoltativo alla procedura centralizzata quando il ricorso a una procedura unica porti netti vantaggi ai pazienti come nel caso di medicinali che rappresentano innovazioni terapeutiche o medicinali non innovativi che possono essere utili alla società o ai pazienti.
La procedura centralizzata ha inizio con la presentazione, da parte dell’azienda farmaceutica, della domanda di autorizzazione all’EMA a cui fa seguito, entro 210 giorni dal deposito del dossier, il rilascio del parere del CHMP (Committee for Medicinal Products for Human Use). In caso di parere positivo la decisione viene ratificata dalla Commissione Europea e pubblicata in Gazzetta Ufficiale dell’Unione Europea (GUUE) (Figura 1).

Figura 1. Iter approvativo di un nuovo farmaco dalla domanda di autorizzazione in EMA con procedura centralizzata alla disponibilità a livello locale italiano
In Italia
Mentre nella fase di approvazione europea l’EMA valuta l’efficacia, la sicurezza e la qualità del farmaco, a livello nazionale l’AIFA deve valutare il place in therapy della terapia nel contesto italiano, il rapporto di costo-efficacia e la sostenibilità per il SSN. Per ottenere la classificazione ai fini della rimborsabilità i farmaci autorizzati a livello europeo vengono pertanto sottoposti alla procedura negoziale con AIFA, che ha inizio con la presentazione del dossier di prezzo e rimborso (P&R).
Strumenti di Early Access
- Legge 189/2012: prevede che un nuovo farmaco, entro 60 giorni dalla pubblicazione dell’AIC comunitaria in GUUE, venga classificato in classe Cnn e possa essere commercializzato prima della conclusione della procedura negoziale in AIFA.
Criticità. La classificazione Cnn, nata con l’intento di rendere immediatamente disponibile un farmaco dopo l’approvazione in Europa, nei fatti spesso si è tradotta in un ostacolo all’accesso in quanto, non prevedendo il rimborso da parte del SSN, l’acquisto del farmaco è a totale carico del cittadino o delle singole strutture ospedaliere o ASL. - Legge 98/2013: stabilisce carattere prioritario per i farmaci orfani, quelli di eccezionale rilevanza terapeutica o sociale e a uso esclusivo ospedaliero e prevede che per questi farmaci l’azienda possa presentare il dossier P&R subito dopo la pubblicazione in GUUE, rinunciando all’inserimento in Cnn. Ha inoltre previsto che in questi casi l’iter negoziale debba terminare entro 100 giorni dalla presentazione della domanda.
Dopo la presentazione del dossier di P&R la Commissione Tecnico Scientifica (CTS) determina il rapporto di costo-efficacia ed esprime un parere, non vincolante, sulla classificazione del farmaco ai fini della rimborsabilità. In seguito a parere positivo della CTS, il Comitato Prezzo e Rimborso (CPR) stabilisce il valore economico del farmaco e imposta la contrattazione con l’azienda. Al termine della contrattazione, dopo aver raggiunto l’accordo su prezzo, classe di rimborsabilità ed eventuali condizioni contrattuali di accesso (registro di monitoraggio, piano terapeutico, MEA – Managed Entry Agreement), il Consiglio di Amministrazione (CdA) di AIFA ratifica la decisione e la Determina di prezzo e rimborso viene pubblicata in GU (Figura 1).
A questo punto, perché il farmaco sia accessibile ai pazienti è necessaria un’ulteriore valutazione a livello regionale. In accordo con la Legge 405/2001, che affida alle Regioni il mandato di attuare le iniziative volte al contenimento dei tetti di spesa, le realtà locali hanno istituito il Prontuario Terapeutico Regionale (PTR) e il Prontuario Terapeutico Ospedaliero Regionale (PTOR) nel quale sono inseriti i farmaci prescrivibili all’interno dei presidi ospedalieri regionali. La maggior parte delle Regioni ha adottato un PTOR vincolante, obbligando le strutture a prescrivere unicamente i farmaci presenti nel prontuario regionale, mentre altre Regioni non hanno adottato un PTOR e il farmaco è disponibile per la prescrizione dopo la pubblicazione della Determina AIFA (Figura 1).
Strumenti di Early Access
- Legge 189/2012: stabilisce che i farmaci innovativi o a innovatività condizionata vengano inseriti immediatamente nei Prontuari Terapeutici Regionali.
Tempi di approvazione
Un’analisi sulle tempistiche di approvazione dei farmaci specifica sull’iter approvativo italiano è stata condotta da Lidonnici et al., che hanno quantificato i tempi impiegati per compiere i diversi step necessari affinché la Determina di P&R di un nuovo farmaco venga pubblicata in GU, dopo l’approvazione a livello europeo.
Dopo l’approvazione europea, la durata media dell’iter approvativo in Italia è di 258 giorni
Lo studio ha evidenziato che, dei 190 farmaci approvati in Europa tra gennaio 2015 e gennaio 2018, 85 hanno concluso l’iter approvativo in Italia con pubblicazione in GU della determina di P&R.
I tempi medi dall’approvazione CHMP e la pubblicazione in GUUE e la successiva pubblicazione in GU della classificazione in Cnn sono stati di 62 e 126 giorni, rispettivamente. La durata media dell’iter approvativo, dall’apertura della procedura in CTS alla pubblicazione della determina in GU, è stata invece di circa 258 giorni (73 giorni tra l’apertura della procedura e il parere della CTS, 94 giorni tra il parere della CTS e quello della CPR e 92 tra il parere della CPR e la pubblicazione in GU della determina AIFA) a cui si aggiungono 94 giorni tra l’applicazione della domanda e l’apertura della pratica in CTS, cioè il tempo necessario per la verifica delle informazioni fornite e l’istruzione della pratica.
Come riportano gli Autori, è interessante notare come il parere sul place in therapy del farmaco e sulla relativa rimborsabilità viene rilasciato da AIFA dopo circa 167 giorni dalla presentazione della domanda, tempi relativamente rapidi che potrebbero essere sfruttati per sviluppare un modello di early access in cui il farmaco viene messo a disposizione del SSN prima della contrattazione del prezzo.
Le sottoanalisi dello studio di Lidonnici et al., condotte su diverse tipologie di farmaci, hanno inoltre evidenziato tempistiche minori per la valutazione dei farmaci innovativi, rispetto ai non innovativi (196 vs 272 giorni) e tempi medi superiori per i farmaci orfani, rispetto ai non orfani (280 vs 251) e per i farmaci oncologici-oncoematologici, rispetto a quelli di altre aree terapeutiche (326 vs 234).
Un’analisi analoga, condotta da Prada et al., si è concentrata sui farmaci oncologici e ha quantificato il tempo trascorso tra la pubblicazione dell’AIC in GUUE e la data del primo acquisto a livello ospedaliero per i farmaci approvati in AIFA tra gennaio 2013 e dicembre 2015. Nel periodo in esame l’EMA ha approvato 34 nuovi farmaci oncologici, 14 dei quali con designazione di farmaco orfano. A luglio 2016, solo 17 erano disponibili sul mercato italiano e il tempo medio di approvazione in AIFA è stato di 248 giorni, contro i 441 giorni necessari per completare l’iter in EMA. A livello europeo non è stata riscontrata particolare differenza di tempistiche tra farmaci orfani e non orfani (450 vs 428 giorni), mentre in Italia la procedura di approvazione è risultata notevolmente più rapida per i farmaci non orfani (335 vs 197 giorni). Un dato interessante riguarda il confronto tra i periodi 2013-2014 e 2015-2016 che ha mostrato una diminuzione da 264 a 219 giorni nel tempo di approvazione in AIFA. A livello regionale i tempi di accesso variano notevolmente da Regione a Regione, passando dai 127 giorni necessari per l’accesso in Piemonte e Valle d’Aosta ai 279 giorni registrati in Basilicata.
Strumenti di Early Access
In Europa
Il Regolamento CE n. 726/2004, per l’istituzione di procedure comunitarie per l’autorizzazione e la sorveglianza dei medicinali per uso umano e veterinari, oltre a stabilire, come visto in precedenza, i criteri per l’accesso alla procedura di approvazione centralizzata, ha anche affermato la necessità di istituire «procedure di valutazione accelerate per i medicinali di maggiore interesse terapeutico e procedure per ottenere autorizzazioni temporanee soggette a condizioni annualmente verificabili». Per attuare le disposizioni del Regolamento 726/2004 l’EMA ha adottato diversi strumenti per accelerare le procedure di autorizzazione dei nuovi farmaci.
Accelerated Assessment
La procedura di valutazione accelerata riduce i tempi di valutazione della domanda di autorizzazione da 210 giorni a 150 o meno. La richiesta deve essere effettuata circa 2-3 mesi prima dell’applicazione della domanda ed è applicabile per i farmaci di maggior interesse per la salute pubblica in particolare sotto il punto di vista dell’innovatività.
Conditional Marketing Authorisation (CMA)
La richiesta di autorizzazione condizionata permette di ottenere l’AIC in tempi più brevi anche in assenza di dati clinici completi e con il vincolo di presentare dati a supporto per consolidare il profilo di efficacia e sicurezza. La Conditional Marketing Authorisation può essere richiesta per farmaci orfani, farmaci per trattare situazioni di emergenza o farmaci per il trattamento di malattie che mettono in pericolo la vita. In generale i dati a disposizione devono dimostrare che i benefici derivanti dalla disponibilità immediata del farmaco sono maggiori dei rischi correlati ai dati clinici mancanti. L’autorizzazione condizionata ha validità di 1 anno e può essere rinnovata annualmente. Può essere convertita in AIC permanente dopo presentazione dei dati mancanti.
Marketing Authorisation under Exceptional Circumstances (UEC)
L’AIC in circostanze eccezionali viene concessa ai quei farmaci per i quali non è possibile ottenere, neanche dopo l’autorizzazione, dati completi a supporto di efficacia e sicurezza in normali condizioni di utilizzo in quanto indicati per patologie molto rare o per ragioni etiche. Questa tipologia di autorizzazione ha validità per 5 anni ed è rinnovabile, ma il rapporto beneficio/rischio viene rivalutato annualmente dal CHMP e generalmente non viene convertita in AIC standard.
In Italia
In Italia sono stati introdotti diversi strumenti normativi per anticipare l’accesso nel mercato italiano di farmaci approvati in Europa.
Legge 648/1996
La Legge n. 648 del 23 dicembre 1996 prevede la possibilità di erogare, a carico del SSN e previo parere vincolante della CTS, farmaci per patologie per le quali non siano ancora disponibili alternative terapeutiche valide e che siano:
- innovativi e già in commercio in altri Stati, ma non ancora autorizzati in Italia;
- non ancora autorizzati e sottoposti a sperimentazione clinica;
- disponibili in Italia, ma approvati con un’indicazione terapeutica diversa.
Inoltre, la Legge n. 79 del 2014 ha esteso l’applicazione di questa norma anche in presenza di alternative terapeutiche valide per medicinali da impiegare per un’indicazione terapeutica diversa da quella autorizzata purché tale indicazione sia nota e conforme a ricerche condotte nell’ambito della comunità medico-scientifica nazionale e internazionale, secondo parametri di economicità e appropriatezza.
Infine, il provvedimento della Commissione Unica del Farmaco del 20 luglio 2000 ha specificato la necessità di studi clinici di fase II affinché un farmaco possa essere inserito nell’elenco previsto dalla Legge 648/1996.
Fondo AIFA del 5%
La Legge n. 326 del 24 novembre 2003 ha istituito un fondo nazionale alimentato dalle aziende farmaceutiche con un contributo pari al 5% delle spese annualmente sostenute per le attività di promozione dirette al medico. Il 50% di tale fondo è destinato alle strutture sanitarie del SSN per l’acquisto di farmaci orfani o di farmaci non ancora autorizzati, ma che rappresentano una speranza di cura per gravi patologie. Le richieste di accesso al fondo fanno riferimento al singolo paziente e possono essere inoltrate dalle Regioni, dai centri di riferimento che hanno in cura i pazienti o da strutture specialistiche individuate dalle Regioni.
- Criticità. Il Fondo AIFA 5% è attualmente poco utilizzato: nel 2017 sono stati erogati € 13,5 milioni su una disponibilità di circa € 17,8 milioni.
- Nuove proposte. Dall’istituzione, nel 2003, del fondo AIFA 5% la ricerca medica ha fatto notevoli passi avanti e oggi sono disponibili nuove terapie per il trattamento di forme rare di tumore che, sebbene non siano ancora rimborsate, hanno a loro supporto robuste evidenze di efficacia. Una revisione della Legge 326/2003 dovrebbe aggiornare i criteri di accessi al fondo includendo, accanto ai farmaci orfani, anche i trattamenti per le forme rare di tumore [AIOM, 2018].
Utilizzo off-label
La Legge n. 94 del 8 aprile 1998 consente la prescrizione, da parte del medico che se ne assume esclusiva e diretta responsabilità, di un farmaco già commercio per l’uso al di fuori delle indicazioni terapeutiche o modalità di somministrazione per il quale è stato approvato. L’utilizzo off-label è rimborsato dalla Regione ed è consentito per il singolo paziente nel caso in cui si ritenga che non possa essere adeguatamente trattato coi farmaci a disposizione per quella determinata indicazione terapeutica.
Uso compassionevole
Il Decreto Ministeriale del 7 settembre 2017 stabilisce i criteri e le modalità per l’uso compassionevole di farmaci:
- non ancora autorizzati in Italia, ma soggetti a sperimentazione clinica (in particolare, il farmaco deve essere oggetto di studi di fase II o III, o di fase I in caso di malattie rare e tumori rari. Inoltre, devono essere disponibili dati sufficienti per formulare un giudizio favorevole su efficacia e tollerabilità);
- provvisti di AIC per indicazioni diverse;
- autorizzati in altri paesi, ma non ancora in Italia.
Il farmaco può essere impiegato per uso compassionevole, e pertanto fornito a titolo gratuito da parte dell’azienda, per il trattamento di malattie rare, di pazienti gravi, in pericolo di vita, oppure affetti da malattie per le quali non esistano alternative terapeutiche valide.
Managed Entry Agreements (MEA)
La necessità di accelerare i tempi di approvazione delle nuove terapie, al fine di permettere il rapido accesso sul mercato, si scontra con le esigenze dei servizi sanitari di contenere la spesa e di garantire l’efficacia e la sicurezza del trattamento. Gli strumenti per garantire l’appropriatezza terapeutica e la sostenibilità economica adottati da AIFA sono gli accordi di accesso condizionato (MEA) e i registri di monitoraggio a supporto di tali schemi. I registri di monitoraggio permettono di superare le incertezze derivanti dalla mancanza di evidenze del farmaco nel real-world, permettendo il monitoraggio dell’appropriatezza prescrittiva e la raccolta di dati epidemiologici e di sicurezza. I dati raccolti mediante il registro di monitoraggio permettono infine di gestire i MEA negoziati in fase di approvazione.
I MEA possono essere accordi di tipo finanziario o basati sulla performance del farmaco (Figura 2). In generale, oltre a permettere un più rapido accesso al mercato per i nuovi farmaci, i MEA consentono al SSN di condividere con l’azienda il rischio derivante dall’assenza di dati real-world e di limitare pertanto l’impatto sulla spesa farmaceutica.
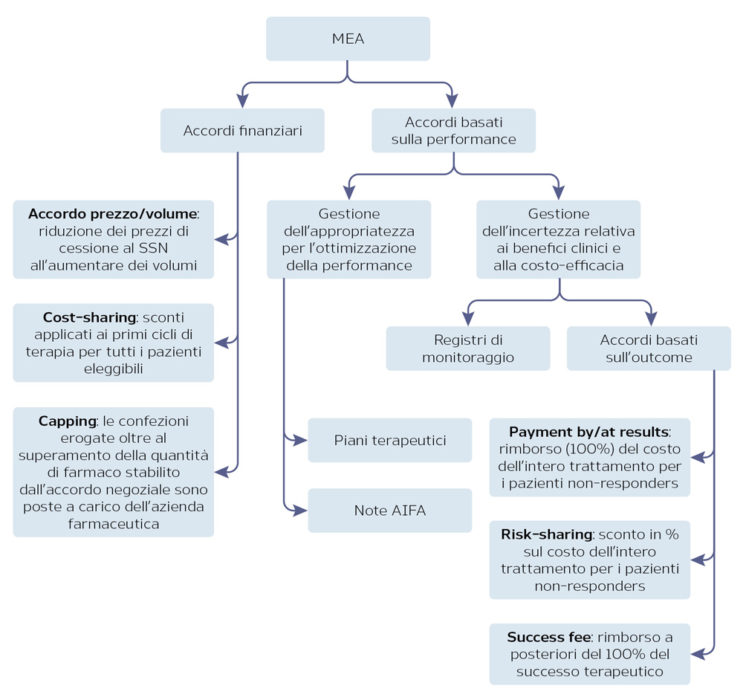
Figura 2. I MEA in Italia
Fonte: AIFA
In merito all’utilizzo dei MEA, lo studio di Villa et al., nel quantificare i tempi di accesso ai farmaci, ha anche indagato l’impatto di strumenti di early access applicati a livello europeo (Conditional Marketing Authorisation – CMA e Marketing Authorisation under Exceptional Circumstances – UEC) sulla procedura di approvazione italiana, in particolare quanto l’incertezza derivante dal processo di autorizzazione a livello centrale abbia influenzato il processo di rimborsabilità a carico del SSN e il ricorso a MEA.
L’analisi ha considerato 65 farmaci che sono stati autorizzati in Europa con CMA (n. 35) o UEC (n. 30) tra luglio 2006 e febbraio 2017. Dei 35 farmaci approvati in Europa con CMA, 28 sono disponibili in Italia e 18 (64%) hanno ottenuto la rimborsabilità in classe A o H, mentre dei 30 autorizzati con UEC, 19 sono disponibili in Italia e 13 (68%) sono stati inseriti in classe A o H. L’analisi dei dati ha inoltre mostrato come l’approvazione a livello europeo mediante CMA sia predittiva di un successivo MEA al momento della negoziazione in AIFA: infatti, dei 18 farmaci approvati con CMA che hanno ottenuto la rimborsabilità in Italia, a 11 (61%) è stato applicato un MEA (5 cost-sharing, 5 payment by results e 1 risk sharing), mentre nessun MEA è stato applicato ai farmaci approvati con UEC.
Per quanto riguarda le tempistiche di approvazione, dal rilascio del parere della CHMP alla pubblicazione della determina di P&R, la procedura è più rapida per i farmaci approvati con CMA (636 vs 897 giorni), tuttavia considerando solo l’iter nazionale (dall’applicazione della domanda di P&R alla pubblicazione in GU) i farmaci autorizzati con UEC presentano tempi più rapidi (329 vs 510 giorni).
Infine, l’introduzione nell’elenco previsto dalla Legge 648/96, richiesto per 3 dei 18 farmaci approvati con CMA e per 6 dei 19 approvati con UEC, ha velocizzato la procedura di approvazione (195 vs 548 giorni per i farmaci CMA e 313 vs 713 per i farmaci UEC), ma ha rallentato l’iter di negoziazione di P&R (805 vs 548 giorni per i farmaci CMA e 818 vs 713 per i farmaci UEC).
Nel mondo
In Tabella I sono riportati alcuni esempi di schemi di early access applicati in USA e in diversi paesi europei.
|
Paese |
Agenzia regolatoria |
Programma |
Caratteristiche |
|
USA |
Food and Drugs Administration (FDA) |
Expanded Access Program (EAP) |
Permette ai pazienti con malattie croniche o in pericolo di vita, per i quali non sono disponibili alternative terapeutiche, di accedere a un farmaco sperimentale al di fuori dagli studi clinici |
|
Francia |
Agence Nationale de Sécurité du Medicament et des Produits de Santé (ANSM) |
Autorisations Temporaires d’Utilisation (ATU) |
Permette l’utilizzo di farmaci non ancora approvati e non soggetti a sperimentazione clinica per il trattamento di malattie gravi o rare per le quali non esiste un’alternativa terapeutica adeguata. Può essere di coorte se l’utilizzo è destinato a gruppi o sottogruppi di pazienti trattati e monitorati secondo criteri definiti in un protocollo terapeutico; sia la prescrizione che la fornitura sono limitate agli ospedali, che negoziano direttamente le condizioni iniziali di “tariffazione gratuita” con l’azienda e il rimborso completo è fornito dal sistema sanitario nazionale. L’ATU nominativo è riferito al singolo paziente e riguarda farmaci il cui rapporto efficacia/sicurezza è ritenuto favorevole alla specifica condizione |
|
Germania |
Das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) |
Arzneimittel-Härtefall-Verordnung (AMHV) |
Consente la distribuzione di farmaci non ancora autorizzati, con trattative di rimborso valutate caso per caso |
|
UK |
Medicines and Healthcare products Regulatory Agency (MHRA) |
Early access to medicines scheme (EAMS) |
Permette l’accesso a farmaci non ancora autorizzati per il trattamento di condizioni potenzialmente letali o gravemente debilitanti in presenza di evidente unmet medical need. L’agenzia regolatoria valuta il rapporto benefici/rischi del farmaco che viene completamente rimborsato |
Tabella I. Esempi di schemi di early access in altre nazioni
Alcune proposte di miglioramento dei programmi di Early Access in Italia
Farmaci orfani
Una serie di proposte per il miglioramento del quadro normativo che regola l’accesso anticipato ai farmaci orfani arriva da un Position Paper pubblicato nel 2018 dall’Osservatorio Farmaci Orfani (OSSFOR) e che ha identificato soluzioni a breve, medio e lungo termine per migliorare l’applicazione della normativa esistente (Legge 648/1996 e Legge 326/2003), ridurre i tempi di approvazione e permettere l’accesso da parte del paziente in tempi più rapidi. In generale, le principali criticità riscontrate riguardano la necessità di un dialogo precoce tra i soggetti coinvolti nei processi e maggiore condivisione delle informazioni.
Le proposte a breve termine comprendono l’agevolazione del confronto tra AIFA e aziende per la verifica della presenza dei presupposti per l’erogazione del farmaco stesso, l’istituzione di un registro delle autorizzazioni all’accesso al fondo AIFA 5% con elenco dei farmaci erogati e relative patologie e il ripristino del rimborso diretto di AIFA alle aziende. Nel medio periodo si propone la creazione di un registro di monitoraggio ad hoc per i dati clinici ed economici dei farmaci inseriti nell’elenco previsto dalla Legge 648/96 e la pubblicazione del procedimento seguito da AIFA per l’autorizzazione all’erogazione dei fondi AIFA 5%. Inoltre, sempre nel medio termine, potrebbero essere superate le criticità associate all’importazione dei farmaci mediante l’emanazione di indicazioni specifiche e linee guida da parte di AIFA.
La normativa italiana può contare su diversi strumenti di early access
Infine, le soluzioni di lungo termine, che riguardano interventi sul piano normativo, suggeriscono la modifica della Legge 648/96 per permettere il coinvolgimento delle aziende in fase di richiesta di inserimento nella lista, la disciplina dei prodotti a cui viene negato il rimborso, la predisposizione di un fondo di riferimento aggiornato annualmente, l’istituzione di un fondo dedicato ai farmaci off-label e la riduzione dell’incertezza sui tempi di negoziazione.
Farmaci oncologici
L’accesso tempestivo alle nuove terapie antitumorali con un significativo valore aggiunto rappresenta una necessità importante per i pazienti e con questo obiettivo un panel di esperti formato da medici oncologi, rappresentanti delle istituzioni, delle aziende farmaceutiche, delle associazioni di pazienti e dei media ha avanzato delle proposte per migliorare gli strumenti di early access a oggi disponibili in Italia [Apolone et al. 2019].
La prima proposta riguarda la definizione dei farmaci meritevoli di early access limitando l’accesso ai farmaci già valutati in EMA con procedura accelerata in quanto ritenuti innovativi e ai nuovi farmaci il cui medical need e valore aggiunto venga valutato secondo criteri ben definiti basati sui criteri di innovatività considerati da AIFA, sulle raccomandazioni all’uso dei farmaci elaborate dall’Associazione Italiana Oncologia Medica (AIOM) e sui criteri per la stratificazione del grado di beneficio clinico dei farmaci della European Society for Medical Oncology (ESMO).
I setting di applicazione di tali criteri sono quello curativo e non curativo, nel caso di malattia in stadio molto avanzato.
I farmaci che otterranno un punteggio superiore alla soglia definita verranno definiti first in class e potranno accedere al percorso di early access che prevede la possibilità di inoltrare la richiesta in AIFA subito dopo il parere positivo del CHMP. Il tempo concesso alla CTS per la valutazione del dossier e l’emissione del giudizio è di 60 giorni dopo il quale, in caso di parere positivo, il farmaco potrà entrare sul mercato ed essere completamente rimborsato dal SSN a un prezzo negoziato in via temporanea e con apposito registro di patologia. Dopo al massimo 12 mesi, al termine della procedura di P&R l’azienda dovrà restituire l’eventuale differenza tra il prezzo definito per l’early access e quello infine negoziato. In caso di rimborsabilità negata il farmaco perde la rimborsabilità e, mentre ai pazienti verrà garantita la continuità terapeutica, l’azienda dovrà rimborsare la spesa sostenuta dal SSN.
I farmaci che, secondo il punteggio di valore clinico, verranno definiti second in class potranno accedere al percorso di early access solo in presenza di evidenze scientifiche che ne dimostrino il beneficio clinico aggiuntivo.
Infine, secondo il panel di esperti l’early access dei farmaci oncologici potrebbe essere finanziato dalla quota non erogata del fondo AIFA 5%, dai rimborsi che l’azienda dovrà eventualmente versare come differenza di prezzo e dal pay back della spesa sostenuta in caso di non rimborsabilità. Nel caso in cui al farmaco venga riconosciuta l’innovatività, il percorso di early access si fermerebbe e il farmaco potrà accedere ai fondi previsti per i farmaci oncologici innovativi.
Bibliografia di riferimento
- Apolone G, Ardizzoni A, Buzzetti G, et al. Early Access in Oncology: why is it needed? Global & Regional Health Technology Assessment 2019; 2019: 1-7
- Associazione Italiana di Oncologia Medica – AIOM. Early access in Italia. Le regole che definiscono l’accesso anticipato ai farmaci anticancro. AIOM, 2018
- Decreto 7 settembre 2017. Disciplina dell’uso terapeutico di medicinale sottoposto a sperimentazione clinica. GU Serie Generale n.256 del 02-11-2017
- IQVIA. EFPIA Patient W.A.I.T. Indicator 2018 survey. EFPIA, 2019 Disponibile su www.efpia.eu/media/412747/efpia-patient-wait-indicator-study-2018-results-030419.pdf (ultimo accesso settembre 2019)
- Legge 23 dicembre 1996, n. 648. Conversione in legge del decreto-legge 21 ottobre 1996, n. 536, recante misure per il contenimento della spesa farmaceutica e la rideterminazione del tetto di spesa per l’anno 1996. GU Serie Generale n.300 del 23-12-1996
- Legge 24 novembre 2003, n. 326. Conversione in legge, con modificazioni, del decreto-legge 30 settembre 2003, n. 269, recante disposizioni urgenti per favorire lo sviluppo e per la correzione dell’andamento dei conti pubblici. GU Serie Generale n.274 del 25-11-2003 – Suppl. Ordinario n. 181
- Legge 8 aprile 1998, n. 94. Conversione in legge, con modificazioni, del decreto-legge 17 febbraio 1998, n. 23, recante disposizioni urgenti in materia di sperimentazioni cliniche in campo oncologico e altre misure in materia sanitaria. GU Serie Generale n.86 del 14-04-1998
- Legge 8 novembre 2012, n. 189 . Conversione in legge, con modificazioni, del decreto-legge 13 settembre 2012, n. 158, recante disposizioni urgenti per promuovere lo sviluppo del Paese mediante un più alto livello di tutela della salute. GU Serie Generale n.263 del 10-11-2012 – Suppl. Ordinario n. 201
- Lidonnici D, Ronco V, Isernia M, et al. Tempi di accesso ai farmaci in Italia nel periodo 2015-2017: Analisi delle tempistiche di valutazione dell’Agenzia Italiana del Farmaco. Global & Regional Health Technology Assessment 2018; 2018: 1-9
- Osservatorio Farmaci Orfani – OSSFOR. Position Paper OSSFOR. Early Access Programmes: Le nostre proposte di miglioramento nell’applicazione della normativa. OSSFOR, 2018. Disponibile su www.osservatoriofarmaciorfani.it/wp-content/uploads/2018/09/OSSFOR_POSITION_PAPER_1_Digitale.pdf (ultimo accesso settembre 2019)
- Prada M, Ruggeri M, Sansone C. Timeline of Authorization and Reimbursement for Oncology Drugs in Italy in the Last 3 Years. Medicine Access @ Point of Care 2017; 1: e29-36
- Provvedimento Commissione Unica del Farmaco 20 luglio 2000. Istituzione dell’elenco delle specialità medicinali erogabili a totale carico del Servizio sanitario nazionale ai sensi della legge 648/96. GU n. 219 del 19-09-2000
- Regolamento (CE) n. 726/2004 del Parlamento europeo e del Consiglio del 31 marzo 2004 che istituisce procedure comunitarie per l’autorizzazione e la sorveglianza dei medicinali per uso umano e veterinario, e che istituisce l’agenzia europea per i medicinali. Gazzetta ufficiale n. L 136 del 30/04/2004. Disponibile su https://eur-lex.europa.eu/legal-content/IT/TXT/HTML/?uri=CELEX:32004R0726&from=IT (ultimo accesso settembre 2019)
- Villa F, Jommi C, Genazzani A, et al. Accesso precoce al mercato: dalle approvazioni condizionate di EMA agli accordi negoziali particolari di AIFA. Global & Regional Health Technology Assessment 2018; 2018: 1-10


