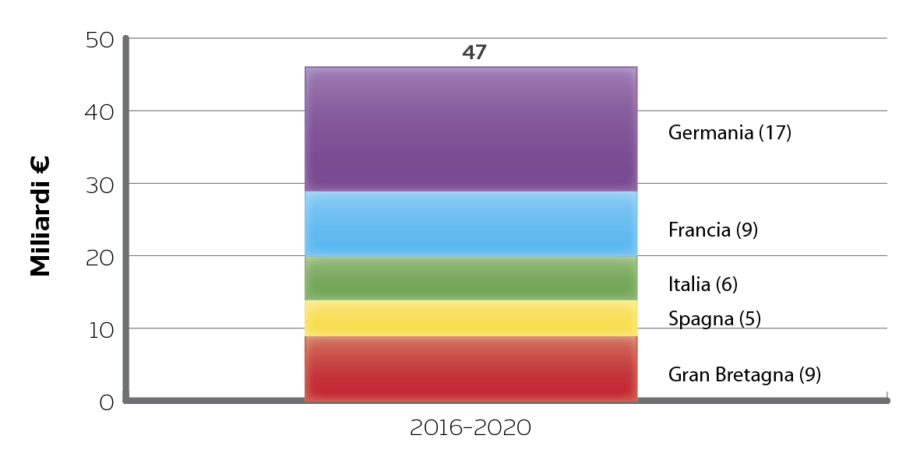
Breve intervista doppia a Loredano Giorni (Regione Piemonte) e Paolo Stella (Regione Puglia)
Il comma 407 della legge di bilancio 2017 contiene alcune novità in merito all’utilizzo dei biosimilari e alle relative procedure pubbliche di acquisto. Obiettivo principale delle disposizioni è la «razionalizzazione della spesa per l’acquisto di farmaci biologici a brevetto scaduto e per i quali siano presenti sul mercato i relativi farmaci biosimilari». Ma che cosa ne pensano i responsabili regionali delle risorse farmaceutiche, ai quali spetta il compito di applicare nella pratica queste procedure e verificarne la validità e i punti critici? L’abbiamo chiesto a Loredano Giorni, Responsabile del settore Assistenza Farmaceutica, Integrativa e Protesica della Regione Piemonte e a Paolo Stella, Dirigente del Servizio Politiche del Farmaco della Regione Puglia.
Intervista a Loredano Giorni
 Secondo lei, le novità sui biosimilari introdotte con la legge di bilancio 2017 possono rappresentare un vantaggio per la sanità pubblica?
Secondo lei, le novità sui biosimilari introdotte con la legge di bilancio 2017 possono rappresentare un vantaggio per la sanità pubblica?
A mio parere, il comma 407, che va a regolamentare le gare, il commercio e la prescrizione dei biosimilari, non comporta particolari vantaggi per la spesa pubblica. Anzi, come parte della pubblica amministrazione, posso dire che non ne sentivamo la necessità, che invece, evidentemente, era sentita da parte di Farmindustria e dalle Associazioni di biosimilari e di generici. Comunque, rispetto a quello che si era prospettato, l’ex articolo 59 della proposta di legge, emendato in base alle correzioni volute dalla Conferenza dei Presidenti delle Regioni, nella sua definitiva stesura ci consente di indire le gare sui farmaci biologici e di ottenere delle significative economie. Insomma, non siamo entusiasti ma neanche preoccupati. Diciamo che manteniamo alta l’attenzione.
Come valuta l’utilizzo di accordi-quadro per la procedura pubblica di acquisto, quando i medicinali biologici con medesima molecola sono più di tre?
Per valutare correttamente la questione degli accordi-quadro, bisognerà vedere come viene interpretata nelle singole realtà. Dal mio punto di vista, come Regione Piemonte, non ritengo che il ricorso ad accordi-quadro possa creare particolari difficoltà. In pratica, la legge di stabilità afferma l’obbligo di stipulare accordi-quadro con le aziende produttrici quando sul mercato sono presenti più di 3 biosimilari dello stesso farmaco originatore. In questo modo diventano fornitori del prodotto le aziende che presentano le offerte economicamente più vantaggiose. Inizialmente la proposta di legge, sempre l’ex articolo 59, prevedeva che il medico non dovesse giustificare la propria prescrizione in nessun caso e in nessun modo. La conversione del disegno di legge ha cancellato questo passaggio e anzi, nella stesura definitiva, prevede che gli enti preposti non solo possono ma devono chiedere al medico una giustificazione basata su evidenze scientifiche allorquando effettui la prescrizione di un biosimilare diverso da quelli frutto dell’accordo-quadro e, in particolare, nel caso di prescrizione nel prodotto che non risulta primo aggiudicatario. In questo modo la legge ci consente di governare il sistema in modo efficace ed economicamente sostenibile.
Per quanto riguarda il limite dei 3 medicinali tra cui scegliere, io sarei stato maggiormente “garantista” e avrei concesso la possibilità di scegliere tra tutti quelli in commercio, sempre a patto, però, di rispettare il vincolo della migliore proposta economica. In sostanza, il criterio applicato sarebbe rimasto comunque lo stesso in modo da poter valutare le decisioni del medico e tenere sotto controllo la spesa sanitaria.
La questione del lotto unico può comportare delle limitazioni all’accesso o dei vincoli al mercato?
Con il lotto unico si prevede che, laddove si tratti di biosimilari, non sia possibile considerare l’equivalenza terapeutica tra molecole diverse ma si debba fare riferimento ad un unico principio attivo, con lo stesso dosaggio e la stessa via di somministrazione. Tutto sommato, questo vincolo non risulta di ostacolo in quanto, comunque, ci muoviamo già all’interno di un sistema concorrenziale che ci consente di governare i prezzi. Anche su questo punto, avremmo fatto volentieri a meno di questo vincolo, ma rispetto alla prima stesura della proposta di legge, che non consentiva gare omogenee per nessun tipo di farmaco, neanche per quelli non biosimilari, la versione definitiva della norma ci lascia comunque dei margini per mettere in concorrenza le aziende produttrici e, in conclusione, governare la spesa.
Nel panorama italiano l’utilizzo dei biosimilari non è uniforme in tutte le Regioni: a suo parere, la legge di bilancio può centrare l’obiettivo di incentivarne il ricorso?
In effetti la penetrazione dei biosimilari nel mercato italiano è davvero molto variegata: secondo stime recenti, si passa da percentuali molto alte, come il 70% della Regione Piemonte, a percentuali che sfiorano appena il 10% in altre Regioni. In questo senso, io mi auguro che la legge di bilancio possa essere uno stimolo e un incentivo verso l’utilizzo dei biosimilari anche per quelle Regioni che al momento risultano più indietro nella loro adozione, e mi sembra chiaro che sia anche questo l’obiettivo del comma 407 laddove afferma che «in caso di scadenza del brevetto o del certificato di protezione complementare di un farmaco biologico durante il periodo di validità del contratto di fornitura, l’ente appaltante entro sessanta giorni dal momento dell’immissione in commercio di uno o più farmaci biosimilari contenenti il medesimo principio attivo, apre il confronto concorrenziale tra questi e il farmaco originatore». Questo vincolo rappresenta un vantaggio per la pubblica amministrazione e si riflette in un incentivo verso il biosimilare.
A questo proposito, mi preme sottolineare un aspetto interessante che noi, come Regione Piemonte, abbiamo inserito nella procedura di gara per la fornitura dei farmaci: se nel periodo di validità di una gara viene immesso sul mercato un farmaco generico o biosimilare, il cui prezzo ex-factory è inferiore al prezzo di aggiudicazione, in attesa di un nuovo bando, all’azienda produttrice che ha vinto la gara viene riconosciuto il prezzo massimo ex-factory del farmaco generico o biosimilare appena autorizzato. In questo modo è possibile per la Regione ottenere un vantaggio economico in base alle regole di mercato e della concorrenza. Questa procedura ci ha causato delle contestazioni da parte di alcune aziende aggiudicatarie, ma siamo in fase di risoluzione dei contenziosi con l’obiettivo di chiuderli favorevolmente per tutti i soggetti coinvolti.
Infine, vorrei evidenziare un aspetto importante sul tema e cioè l’impiego dei biosimilari nei pazienti naïve. Alcuni ritengono che il farmaco biosimilare possa essere prescritto solo ed esclusivamente nel paziente naïve, ma non esistono dati scientifici a supporto di questa affermazione. Nella Regione Piemonte abbiamo eliminato questa distinzione e abbiamo disposto che il medico debba presentare una giustificazione scientifica in caso prescriva un farmaco diverso da quello che si è aggiudicato con gara non solo per il paziente naïve ma anche per quello già in trattamento con il farmaco biotecnologico originatore. Questa disposizione è stata messa in atto, ad esempio, meno di un anno fa in relazione al biosimilare dell’insulina glargine: oggi in varie ASL della nostra Regione il consumo del farmaco biosimilare, aggiudicatario della gara, supera l’80% del consumo totale di insulina glargine con punte di oltre il 95%. È evidente che non possa trattarsi di soli pazienti naïve ma che ci sia una percentuale per i quali è stato effettuato lo “switch” rispetto al farmaco originatore. In questo modo si è garantita la continuità terapeutica e allo stesso tempo si sono generati notevoli risparmi per poter effettuare nuovi ed efficaci investimenti per la salute dei cittadini. A mio parere questa è la chiave per incentivare la prescrizione del biosimilare e raggiugere le percentuali di utilizzo registrate, ad esempio, nella nostra Regione.
Intervista a Paolo Stella
 Secondo lei, le novità sui biosimilari introdotte con la legge di bilancio 2017 possono rappresentare un vantaggio per la sanità pubblica?
Secondo lei, le novità sui biosimilari introdotte con la legge di bilancio 2017 possono rappresentare un vantaggio per la sanità pubblica?
Non sono sicuro che possano rappresentare un vantaggio per la sanità pubblica, in quanto restringono la possibilità per le aziende sanitarie di svolgere un’attività negoziale tramite procedure ad evidenza pubblica in alcuni casi anche più selettive, limitando in tal modo la convenienza economica. Tuttavia, riconosco che possano consentire di raggiungere risultati economici positivi dal momento che la normativa prevede l’utilizzo di accordi-quadro, che costituiscono anch’essi una procedura di acquisto ad evidenza pubblica e danno l’opportunità a più operatori economici di aggiudicarsi una parte del lotto acquistato. Sebbene io veda una riduzione dal punto di vista del risparmio ottenibile applicando le norme introdotte dalla legge di bilancio rispetto ad una procedura ad evidenza pubblica, convengo che tali norme rappresentino “un male minore” rispetto ad un acquisto effettuato con procedura negoziata diretta con i diversi fornitori: è un modo per risparmiare, ma non è quello più efficace.
A suo parere, quale potrebbe essere il metodo più efficace per innescare un risparmio nella spesa sanitaria pubblica in merito ai farmaci biotecnologici e biosimilari?
In realtà, io ritengo che questo compito, che è stato affidato alle Regioni e alle stazioni appaltanti, sia un compito e una decisione che spetti all’AIFA perché, se l’Agenzia Italiana del Farmaco assume una presa di posizione più chiara in merito, elaborando e pubblicando dei pareri di equivalenza terapeutica effettivi su queste categorie di farmaci, di certo le stazioni appaltanti possono risultare molto più facilitate nell’effettuare procedure di acquisto senza incorrere in contenziosi con i fornitori. Potendo contare su una base normativa più forte e più decisa a livello centrale, sicuramente a livello periferico le Regioni e le stazioni appaltanti potranno essere avvantaggiate nella messa in atto di procedure di acquisto con maggiori livelli di autonomia e, di conseguenza, migliori risultati di economicità.
La questione del lotto unico può comportare delle limitazioni all’accesso o dei vincoli al mercato?
In questo caso la norma, secondo me, contiene una incongruenza di base che può creare notevoli problemi di interpretazione e applicazione. Infatti, se da una parte si afferma che «non possono essere posti in gara nel medesimo lotto principi attivi differenti, anche se aventi le stesse indicazioni terapeutiche», più avanti nella stessa norma si dice che per la costituzione del lotto unico di acquisto «si devono considerare lo specifico principio attivo (ATC di V livello), i medesimi dosaggio e via di somministrazione». Se prendiamo come esempio le eritropoietine, appare chiara l’incongruenza a cui accennavo: infatti, allo stesso ATC di V livello, cioè B03XA01, corrispondono sia l’eritropoietina alfa sia l’eritropoietina beta. Questa situazione può generare confusione nell’elaborazione del bando di gara e comportare l’avvio di ricorsi in giudizio da parte dei diversi operatori economici interessati. Su questo punto, a mio parere, sono necessari dei chiarimenti e degli approfondimenti, per guidare con chiarezza l’operato delle Regioni e delle stazioni appaltanti, ed evitare l’impugnazione dei risultati da parte dei fornitori.
Nel panorama italiano l’utilizzo dei biosimilari non è uniforme in tutte le Regioni: a suo parere, la legge di bilancio può centrare l’obiettivo di incentivarne il ricorso?
Di certo poter fare riferimento ad un indirizzo uguale per tutti rappresenta un vantaggio perché, in qualche modo, consente anche di livellare e uniformare le condizioni economiche a livello regionale e soprattutto tra le Regioni del nord e quelle del sud, dove storicamente esiste un divario notevole. Avere una normativa che preveda procedure comuni per le diverse realtà è importante ma, per trarne il beneficio maggiore, è altrettanto importante che la normativa sia chiara e che possa essere interpretata da tutti alla stessa maniera. Perché, ripeto, non è opportuno che si verifichino casi di diversa interpretazione e applicazione della legge, ad esempio in merito alla definizione del lotto unico da mettere in gara, così come abbiamo visto prima per le eritropoietine. Senza la sufficiente chiarezza, la normativa che ha l’obiettivo di uniformare le strategie a livello delle Regioni rischia di veder limitato il suo valore.
Legge di bilancio 2017. Comma 407
«L’esistenza di un rapporto di biosimilarità tra un farmaco biosimilare e il suo biologico di riferimento sussiste solo ove accertato dalla European Medicine Agency (EMA) o dall’Agenzia italiana del farmaco, tenuto conto delle rispettive competenze. Non è consentita la sostituibilità automatica tra farmaco biologico di riferimento e un suo biosimilare né tra biosimilari. Nelle procedure pubbliche di acquisto per i farmaci biosimilari non possono essere posti in gara nel medesimo lotto principi attivi differenti, anche se aventi le stesse indicazioni terapeutiche. Al fine di razionalizzare la spesa per l’acquisto di farmaci biologici a brevetto scaduto e per i quali siano presenti sul mercato i relativi farmaci biosimilari, si applicano le seguenti disposizioni:
a) le procedure pubbliche di acquisto devono svolgersi mediante utilizzo di accordi-quadro con tutti gli operatori economici quando i medicinali sono più di tre a base del medesimo principio attivo. A tal fine le centrali regionali d’acquisto predispongono un lotto unico per la costituzione del quale si devono considerare lo specifico principio attivo (ATC di V livello), i medesimi dosaggio e via di somministrazione;
b) al fine di garantire un’effettiva razionalizzazione della spesa e nel contempo un’ampia disponibilità delle terapie, i pazienti devono essere trattati con uno dei primi tre farmaci nella graduatoria dell’accordo-quadro, classificati secondo il criterio del minor prezzo o dell’offerta economicamente più vantaggiosa. Il medico è comunque libero di prescrivere il farmaco, tra quelli inclusi nella procedura di cui alla lettera a), ritenuto idoneo a garantire la continuità terapeutica ai pazienti;
c) in caso di scadenza del brevetto o del certificato di protezione complementare di un farmaco biologico durante il periodo di validità del contratto di fornitura, l’ente appaltante, entro sessanta giorni dal momento dell’immissione in commercio di uno o più farmaci biosimilari contenenti il medesimo principio attivo, apre il confronto concorrenziale tra questi e il farmaco originatore di riferimento nel rispetto di quanto prescritto dalle lettere a) e b);
d) l’ente appaltante è tenuto ad erogare ai centri prescrittori i prodotti aggiudicati con le procedure previste dal decreto legislativo 18 aprile 2016, n. 50;
e) eventuali oneri economici aggiuntivi, derivanti dal mancato rispetto delle disposizioni del presente comma, non possono essere posti a carico del Servizio sanitario nazionale»
Secondo lei, la nuova norma garantisce la continuità terapeutica?
Il ricorso alla procedura pubblica di acquisto tramite accordo-quadro tutela la continuità terapeutica, a patto di includere all’interno dell’accordo-quadro tutti gli operatori presenti sul mercato. La legge non pone limiti sul numero di operatori economici da coinvolgere ma specifica solo che, per poter effettuare una procedura di acquisto tramite accordo-quadro, devono esserci almeno 3 medicinali a base del medesimo principio attivo. Quindi, potrebbe darsi l’evenienza che ci siano 4 terapie erogate da 4 operatori diversi, e in questo caso è necessario prevedere, ed è auspicabile, la partecipazione alla gara di tutti gli operatori coinvolti: in caso contrario, per alcuni pazienti, potrebbe non essere garantita la continuità terapeutica.
Inoltre, il meccanismo dell’accordo-quadro potrebbe, in alcuni casi, diventare addirittura un ostacolo al risparmio se ipotizziamo, ad esempio, che tra gli aggiudicatari di un lotto di gara si trovi un farmaco che gode di una importante fetta di mercato e che, pur non avendo avanzato una proposta di prezzo competitiva, continui a vendere “accontentandosi” della fetta di mercato della continuità terapeutica. In questo caso, il possibile risparmio per il mercato, dovuto alla concorrenza, sarebbe molto attenuato.