Il 27 marzo 2018 è stato presentato ufficialmente il Secondo Position Paper AIFA sui Farmaci Biosimilari. Un passo importante e atteso, frutto di lungo lavoro di revisione e aggiornamento del Primo Position Paper, risalente al 2013. Tale revisione è stata condotta anche tramite una consultazione pubblica che, nel corso degli anni, ha coinvolto Società scientifiche, Commissioni regionali, Aziende ospedaliere, Industrie farmaceutiche ma anche Associazioni di pazienti e della società civile.
Un pubblico numeroso di operatori del settore (clinici, figure delle istituzioni e rappresentanti di pazienti) ha partecipato all’incontro dal titolo “Accesso alle terapie con farmaci biologici: i fenomeni di sottotrattamento e le opportunità offerte dai biosimilari”, nel corso del quale Mario Melazzini, Direttore Generale dell’AIFA, e Simona Montilla, Dirigente del Centro studi AIFA, hanno esposto gli obiettivi e le novità di questo Secondo Position Paper. Per rendere conto del contesto e analizzare il possibile sottoutilizzo dei farmaci biologici nella prassi clinica, Luca Minotti, Partner di Ernst&Young per il settore Life Sciences, ha illustrato i risultati di un’indagine condotta per l’Italian Biosimilars Group.
Erano inoltre previsti una serie di Tavoli di lavoro tematici, organizzati da AIFA; oltre a un Tavolo dedicato alle “Opportunità e sfide” sui biosimilari, i diversi esperti sono stati riuniti in Tavoli dedicati alle aree cliniche della Reumatologia, della Gastroenterologia, della Dermatologia e dell’Oncologia.
Un nuovo impegno a tutela della salute dei cittadini
La giornata ha preso il via con l’intervento introduttivo del Direttore Melazzini, che ha ricordato come il 2018 rappresenti un anno di anniversari importanti per la sanità italiana: ricorrono infatti i 40 anni dall’istituzione del Servizio Sanitario Nazionale (Legge 23 dicembre 1978, n. 833) e i 15 anni dell’istituzione dell’Agenzia Italiana del Farmaco (Legge 24 novembre 2003, n.326). Queste date testimoniano l’impegno a favore dei cittadini e dei pazienti, sancito anche dall’articolo 32 della Costituzione italiana, e che viene rinnovato con la pubblicazione del Secondo Position Paper sui biosimilari.
Il Professor Melazzini ha sottolineato come uno dei doveri dell’AIFA sia quello di garantire a tutti i pazienti l’accesso alle cure e pertanto, in un ambito di costante aumento della spesa sanitaria, sia necessario affrontare il tema della sostenibilità del sistema. In quest’ottica, con l’utilizzo dei biosimilari si può fornire un’opzione terapeutica valida e l’accesso a terapie ad alto costo ad un numero maggiore di pazienti. I biosimilari rappresentano inoltre, per il mercato, un elemento di concorrenza che può contribuire al governo della spesa, con la riduzione dei costi non solo sanitari ma anche sociali. In base al Rapporto OSMED 2016, importanti farmaci biologici e biosimilari (come ad esempio trastuzumab, bevacizumab, rituximab, infliximab, adalimumab ed etanercept) figurano tra i primi 30 principi attivi nella spesa regionale per medicinali erogati sia in assistenza farmaceutica ospedaliera e ambulatoriale sia in distribuzione diretta e per conto. Secondo Melazzini, il dibattito sul tema dei biosimilari può costituire un utile esercizio anche in relazione all’ambito dei farmaci innovativi, in quanto si tratta di un’allocazione di risorse importante con notevoli ricadute sulla pratica clinica e un possibile recupero di risorse per il sistema sanitario.
«L’AIFA considera i biosimilari come prodotti intercambiabili con i corrispondenti originatori di riferimento»
Quindi si è passati a presentare nel dettaglio il nuovo Position Paper, che si pone l’obiettivo principale di «fornire agli operatori sanitari e ai cittadini informazioni chiare, trasparenti e convalidate sui medicinali biosimilari», in particolare su 3 aspetti: la definizione dei medicinali biologici e biosimilari, le normative regolatorie vigenti nella UE e il ruolo dei biosimilari nella sostenibilità economica del SSN.
Per la definizione di medicinale biologico il Position Paper fa riferimento a quella elaborata dall’Agenzia europea per i medicinali (EMA), che ha svolto un ruolo di avanguardia nel processo di regolamentazione del processo di autorizzazione dei biosimilari e garantisce un altissimo livello di valutazione pre-immissione in commercio e di monitoraggio post-marketing. Inoltre si sottolineano le caratteristiche che differenziano i farmaci biologici dalle molecole di sintesi chimica, come la dimensione molecolare, la complessità strutturale, il procedimento di derivazione da sistemi diversi. Per tutti questi aspetti, si può affermare che, nel caso dei farmaci biologici, «il prodotto è il processo». Anche per la definizione di medicinale biosimilare si rimanda alla legislazione dell’Unione europea che ha affrontato l’argomento in diversi documenti, a partire dal 2001, fino all’elaborazione di due importanti pubblicazioni nel 2017, una rivolta agli operatori sanitari (“Guida informativa sui medicinali biosimilari”) e una rivolta ai pazienti (Documento di Consenso: “Cosa c’è da sapere riguardo ai Medicinali Biosimilari”). In sintesi, il Position Paper afferma che «un biosimilare e il suo prodotto di riferimento, essendo ottenuti mediante processi produttivi differenti, non sono identici, ma essenzialmente simili in termini di qualità, sicurezza ed efficacia».
Il Direttore Melazzini ha introdotto quindi il tema principale, cioè la posizione di AIFA sui biosimilari come alternativa ai prodotti originatori che, rispetto ai Documenti precedenti (Primo Position Paper del 2013 e Secondo Concept Paper del 2016), fa registrare alcune significative differenze.
Innanzitutto, si conferma che «i medicinali biologici e biosimilari non possono essere considerati sic et simpliciter alla stregua dei prodotti generici, o equivalenti». Nei Documenti precedenti, questa affermazione era seguita da una frase che ribadiva una differenza fondamentale rispetto ai generici, escludendo «la vicendevole sostituibilità terapeutica automatica» (2013) o, più semplicemente, «la sostituibilità automatica» (2016). Inoltre, nel 2013, si specificava anche che l’AIFA aveva deciso di «non includere i medicinali biosimilari nelle liste di trasparenza che consentono la sostituibilità automatica tra prodotti equivalenti». Nel Secondo Position Paper, queste limitazioni vengono eliminate, lasciando scorgere un indizio di cambiamenti ancora maggiori.
«I biosimilari sono uno strumento irrinunciabile per lo sviluppo di un mercato competitivo e concorrenziale che produca salute»
Fondamentale è sempre il ruolo del medico («la scelta di trattamento rimane una decisione clinica affidata al medico prescrittore»), che viene investito anche di un nuovo compito, rispetto ai Documenti precedenti: «contribuire a un utilizzo appropriato delle risorse ai fini della sostenibilità del sistema sanitario e la corretta informazione del paziente sull’uso dei biosimilari».
Quindi, con un richiamo alle normative UE e alle garanzie costituite dalle procedure EMA, si introduce il concetto più innovativo del nuovo Position Paper: «Come dimostrato dal processo regolatorio di autorizzazione, il rapporto rischio-beneficio dei biosimilari è il medesimo di quello degli originatori di riferimento. Per tale motivo, l’AIFA considera i biosimilari come prodotti intercambiabili con i corrispondenti originatori di riferimento». Viene dunque affermata l’intercambiabilità tra farmaco originatore e biosimilare, che finora non era prevista dalle normative italiane e dell’Unione europea. Giova forse ricordare qui che con intercambiabilità si intende la «pratica medica di sostituire un farmaco con un altro, che si prevede produca il medesimo effetto clinico in un determinato contesto clinico in qualsiasi paziente, su iniziativa o con l’accordo del medico prescrittore». Mentre con sostituibilità si intende la «pratica di sostituire un farmaco con un altro farmaco, spesso più economico per il Servizio Sanitario o per il paziente, che abbia la stessa composizione qualitativa e quantitativa di sostanze attive, la stessa forma farmaceutica e via di somministrazione e sia bioequivalente con il medicinale di riferimento sulla base di appropriati studi di biodisponibilità». La sostituibilità automatica rimane un elemento caratterizzante degli equivalenti, e riguarda la facoltà del farmacista di dispensare il generico senza consultare il medico prescrittore.
Un’ulteriore novità sostanziale riguarda i pazienti da indirizzare a trattamento con biosimilari. Nel Position Paper 2013 si specificava che i biosimilari fossero «da preferire, qualora costituiscano un vantaggio economico, in particolare per il trattamento dei soggetti “naïve” (che non abbiano avuto precedenti esposizioni terapeutiche o per i quali le precedenti esposizioni in base al giudizio del clinico siano sufficientemente distanti nel tempo)». Anche questa limitazione decade, nel nuovo Position Paper, che afferma come l’intercambiabilità tra originator e biosimilare «vale tanto per i pazienti naïve quanto per i pazienti già in cura». Un nuovo elemento a supporto dello switch terapeutico, che costituisce un argomento molto dibattuto dai clinici e dalle istituzioni e, recentemente, anche al centro di pronunce giurisprudenziali.
In conclusione del suo intervento, il Direttore Melazzini ha definito i biosimilari come «uno strumento irrinunciabile per lo sviluppo di un mercato competitivo e concorrenziale che produca salute, perché costituiscono un’opzione terapeutica a costo inferiore per il Servizio Sanitario Nazionale (SSN)».
Il quadro scientifico regolatorio in Italia e nell’Unione europea
La presentazione del nuovo Position Paper è proseguita con la Dottoressa Montilla che ha affrontato il tema del quadro scientifico-regolatorio dei farmaci biosimilari, analizzando il processo di autorizzazione che, come abbiamo accennato, ha visto l’Unione europea come «pioniera nel definire un corpus regolatorio, robusto e di alto livello scientifico». Montilla ha illustrato in sintesi i diversi step del processo autorizzativo e i criteri richiesti per gli studi di confronto necessari, ricordando come l’EMA abbia messo a punto diverse linee guida dedicate all’approvazione dei biosimilari, da quelle più generali, a quelle specifiche per il singolo prodotto (ad esempio insulina, somatropina, epoetine), a quelle riguardanti aspetti peculiari dei biosimilari come l’esercizio di comparabilità e l’immunogenicità.
Quindi la Dottoressa Montilla ha presentato il tema dell’estrapolazione delle indicazioni terapeutiche, evidenziando le differenze rispetto al precedente Position Paper: se nel 2013 si diceva che «il Committee for Medicinal Products for Human Use (CHMP) dell’EMA stabilisce caso per caso se le indicazioni multiple possano essere estrapolate sulla base delle evidenze scientifiche derivanti da un esercizio di comparabilità approfondita e in conformità ad opportune giustificazioni scientifiche», il nuovo Position Paper specifica che la valutazione del CHMP deve avvenire non solo «caso per caso» ma anche «preventivamente» e che «tale estrapolazione non è automatica». Inoltre l’AIFA sottolinea come questa pratica rappresenti «un principio scientifico e regolatorio esercitato da molti anni su tutti i farmaci biotecnologici, sia nel caso di importanti variazioni nei processi di produzione, sia nel caso dell’immissione in commercio di nuove formulazioni dello stesso farmaco biotecnologico». Questo discorso si collega alle peculiarità del processo produttivo dei farmaci biologici che comportano l’unicità del prodotto («il prodotto è il processo»), con differenze variabili da un laboratorio ad un altro. Ad esempio, come riportato nel Position Paper, nel caso di trastuzumab sottocute, già in passato è stato richiesto di condurre studi clinici per estrapolare l’indicazione di trastuzumab endovena nel carcinoma mammario (e non si trattava di un’estrapolazione del biosimilare).
«L’estrapolazione è un principio scientifico e regolatorio esercitato da molti anni su tutti i farmaci biotecnologici»
Un altro cambiamento rispetto al precedente Position Paper riguarda le modalità di inserimento dei biosimilari nell’elenco delle specialità erogabili a carico del SSN ai sensi della Legge n. 648/96. In particolare, si specifica che «affinché un farmaco possa essere inserito in tale elenco – e sia dunque a carico del SSN» debbano essere «disponibili adeguati dati di sicurezza ed efficacia raccolti in studi clinici almeno di fase II e, se del caso, che siano posti in essere idonei strumenti di monitoraggio a tutela della sicurezza dei pazienti». Inoltre si precisa che «lo stesso requisito di presenza di studi clinici adeguati almeno di fase II si applica anche per i farmaci impiegati per una indicazione off-label in presenza di alternative terapeutiche autorizzate, ma più onerose per il SSN». L’utilizzo dei farmaci biologici e biosimilari in off-label viene affrontato in maniera molto dettagliata, innanzitutto confermando la possibilità che anche i biologici possano essere utilizzati in off-label e quindi chiamando in causa la Commissione Tecnico Scientifica (CTS) dell’AIFA che deve verificare «caso per caso» il possibile inserimento nell’elenco relativo alla Legge n. 648/96 di un biosimilare il cui originator è già presente in tale elenco. Tale inserimento non risulta automatico e dipende pertanto dalla CTS «che si riserva la possibilità di esprimere il proprio parere sulla base delle evidenze scientifiche e della letteratura disponibili, dell’esperienza clinica e dell’eventuale riconducibilità dell’azione terapeutica ad un identico meccanismo d’azione». In quest’ottica, il Position Paper ricorda i recenti Criteri generali elaborati dalla CTS per procedere a questo tipo di valutazione, le cui basi rimangono le Linee guida EMA per le indicazioni autorizzate: in particolare si richiama la necessità di individuare «tutti gli elementi del comparability exercise riguardanti qualità, preclinica e clinica contenuti nell’EPAR (European Public Assessment Report)» dell’EMA; valutare «se il meccanismo d’azione del farmaco nell’indicazione autorizzata e rimborsata ai sensi della legge n.648/96 sia riconducibile o meno a caratteristiche della molecola diverse da quelle valutate e approvate nel comparability exercise»; se esistono «safety concerns» per l’indicazione richiesta. In ultimo, si specifica che, per farmaci particolari, può essere richiesta la presenza di studi bridging.
L’analisi del quadro normativo è stata quindi completata dalla dottoressa Montilla con la presentazione delle procedure di farmacovigilanza previste a livello italiano e a livello europeo per i farmaci biologici e biosimilari: il Piano di monitoraggio intensivo predisposto da AIFA (contrassegnato dal triangolo equilatero rovesciato riportato sul foglietto illustrativo e sul Riassunto delle Caratteristiche del Prodotto) e il Risk Management Plan richiesto da EMA. Inoltre è stata anche ricordata la procedura di prezzo e rimborso stabilita per il nostro Paese: in base alla Delibera CIPE del 1° febbraio 2001, per tutti i biologici e i biosimilari, così come per gli altri farmaci rimborsabili, viene condotta da AIFA una negoziazione del prezzo con l’azienda produttrice. Nel caso dei generici e dei biosimilari il Decreto 4 aprile 2013 del Ministero della Salute (il cosiddetto “Decreto scaglioni”) prevede la riduzione di prezzo di almeno il 20%, rispetto al prezzo del farmaco originatore.
L’inserimento dei biosimilari nell’elenco della Legge n. 648/96 è verificato «caso per caso» dalla CTS di AIFA
In ultimo, la dottoressa Montilla è tornata sul tema dell’intercambiabilità per tracciare il trend a livello europeo: se da una parte l’EMA si concentra sulle procedure di immissione al commercio dei biologici e dei biosimilari senza fornire indicazioni sul possibile switch, dall’altra le decisioni di alcuni paesi europei, come Francia, Germania, Finlandia e Portogallo, considerano i biosimilari intercambiabili con gli originator, pur sottolineando la necessità di una corretta informazione del paziente e di un costante monitoraggio della terapia. Il Secondo Position Paper AIFA sembra quindi seguire questo trend.
L’analisi del sottotrattamento da biologico
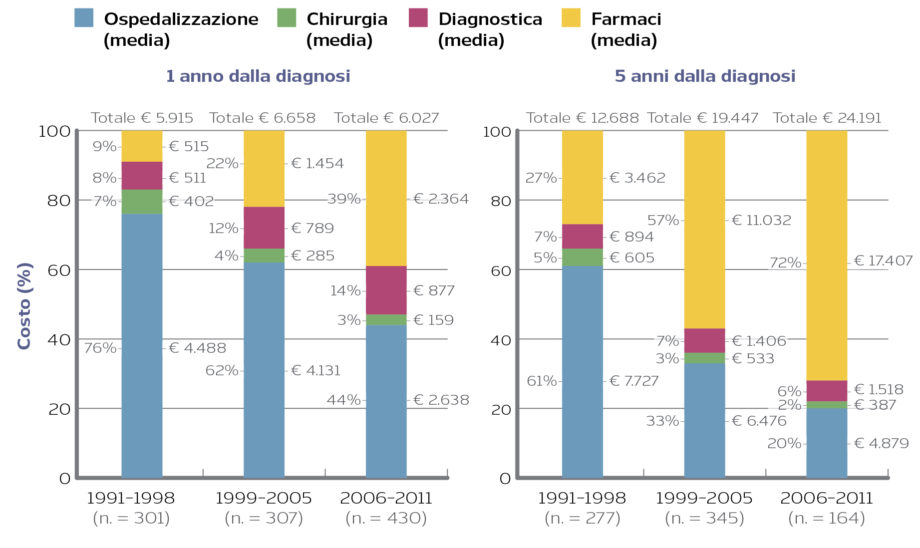
L’incontro del 27 marzo ha anche fornito dati interessanti sulla pratica clinica legata alle terapie biologiche in Italia, grazie alla presentazione dell’analisi condotta da Ernst&Young per l’Italian Biosimilars Group con l’obiettivo di indagare i possibili fenomeni di sottotrattamento da biologico.
Lo studio, illustrato da Luca Minotti, è stato effettuato su 11 patologie per le quali sono già disponibili anche i biosimilari: psoriasi, artrite psoriasica, spondilite anchilosante, artrite reumatoide, malattia di Crohn, colite ulcerosa, linfoma non-Hodgkin, leucemia linfatica cronica, carcinoma mammario, tumore al colon retto e melanoma metastatico. Per ogni patologia sono stati confrontati i dati relativi ai pazienti eleggibili al trattamento con la terapia biologica (in base alle evidenze di letteratura, alle linee guida e alla pratica clinica) e i dati dei pazienti che risultano trattati con i biologici (in base ai dati di mercato IQVIA): l’eventuale differenza riscontrata è stata considerata come condizione di sottotrattamento per la singola patologia.
Nonostante la variabilità dei dati tra le diverse patologie considerate, i risultati complessivi hanno evidenziato che in Italia circa 200.000 pazienti potenzialmente eleggibili non sono trattati con la terapia biologica (con un range da 100.000 a 300.000 pazienti).
In particolare, Minotti ha riportato i numeri relativi ad alcune patologie: per la psoriasi, il range di pazienti eleggibili al trattamento è molto ampio e risente di una importante differenza tra quanto risulta dalla letteratura (200.000 pazienti eleggibili) e il dato riportato dall’esperto (50.000 pazienti eleggibili); di conseguenza, poiché i pazienti in trattamento risultano essere 16.000, il dato dei sottotrattati varia da 184.000 a 34.000. Per l’artrite psoriasica, i pazienti eleggibili sono tra 34.500 e 48.000 e i pazienti trattati sono 24.000, il sottotrattamento riguarda quindi tra 10.500 e 24.000 pazienti. Per la spondilite anchilosante, i pazienti eleggibili sono 29.000, 9.000 sono i trattati e 20.000 i sottotrattati. Per l’artrite reumatoide, i pazienti eleggibili sono 43.000-58.000, con 38.000 trattati e un numero di sottotrattati variabile tra 5.000 e 20.000. Per la malattia di Crohn, i pazienti eleggibili sono 29.000, con 11.000 trattati e 18.000 sottotrattati. Per la colite ulcerosa, i pazienti eleggibili sono compresi tra 15.000 e 49.000, con 8.000 trattati e un numero di sottotrattati compreso tra 7.000 e 41.000.
Circa 200.000 pazienti potenzialmente eleggibili non sono trattati con la terapia biologica
La situazione delle patologie oncoematologiche è risultata peculiare rispetto alle altre in quanto, secondo il parere degli esperti interpellati nel corso dello studio, i pazienti potenzialmente eleggibili vengono trattati con la terapia biologica senza limitazioni di accesso, probabilmente anche in relazione alla gravità della malattia. Come sottolineato da Minotti, quest’area sembra necessitare di ulteriori approfondimenti per una comprensione migliore del fenomeno.
La discussione dei Tavoli tematici
I risultati dell’analisi di Ernst&Young hanno fornito anche lo spunto di partenza per la discussione dei Tavoli di lavoro tematici, che si è svolta nel pomeriggio e ha fatto emergere significative indicazioni degli esperti coinvolti.
Innanzitutto, è stato evidenziato che, nell’analisi di dati di trattamento (e relativo sottotrattamento) è necessario stratificare i pazienti considerando anche le diverse linee terapeutiche.
In generale, in tutte le aree terapeutiche, è stato segnalato un gap informativo, con la richiesta di implementare flussi amministrativi regionali e nazionali in grado di monitorare non solo l’appropriatezza ma anche la governance della spesa. Studi epidemiologici, registri di patologia e PDTA (Percorsi Diagnostico-Terapeutici Assistenziali) sono stati individuati dai partecipanti ai Tavoli come utili strumenti di analisi e incentivo al corretto utilizzo.
I Tavoli tematici
Opportunità e sfide: AIFA all’ascolto sui biosimilari
Facilitatori:
Patrizia Popoli (Istituto Superiore di Sanità – Presidente CTS AIFA), Armando Genazzani (Università Piemonte Orientale – Membro CTS AIFA)
Partecipanti:
Tonino Aceti (Coordinatore Nazionale Tribunale per i Diritti del Malato e Responsabile Nazionale CnAMC di Cittadinanzattiva), Giordano Beretta (Presidente Eletto Associazione Italiana di Oncologia Medica – AIOM), Luisa Brogonzoli (Fondazione The Bridge), Mauro Galeazzi (Presidente Società Italiana Reumatologia – SIR), Antonio Gasbarrini (Fondazione Policlinico Universitario A. Gemelli, Roma), Loredano Giorni (Regione Piemonte), Massimo Morosetti (Ospedale Giovan Battista Grassi, Roma), Alessandro Mostaccio (Segretario Generale Movimento Consumatori), Carmine Pinto (UOC Oncologia Medica IRCCS Arcispedale S. Maria Nuova, Reggio Emilia), Luca Minotti (Ernst & Young), Ida Fortino (AIFA)
Area clinica Reumatologia
Facilitatori:
Guido Valesini (Società Italiana Reumatologia – SIR)
Partecipanti:
Antonella Celano (Presidente Associazione nazionale persone con Malattie Reumatologiche e Rare – APMAR), Roberto Gorla (AO Spedali Civili di Brescia), Carlo Maurizio Montecucco (Collegio dei Presidenti Emeriti Società Italiana Reumatologia – SIR), Giuseppe Nocita (SUMAI), Stefano Stisi (Presidente Collegio Reumatologi Italiani – CREI), Silvia Tonolo (Presidente Associazione nazionale Malati Reumatici ONLUS – ANMAR), Francesca Tosolini (Regione Friuli Venezia Giulia), Francesco Deligios (Ernst & Young), Simona Montilla (AIFA)
Area clinica Gastroenterologia
Facilitatori:
Silvio Danese (Centro Malattie Infiammatorie Croniche Intestinali, Humanitas, Rozzano)
Partecipanti:
Domenico Alvaro (Presidente Società Italiana Gastroenterologia, SIGE), Alessandro Armuzzi (Segretario Generale di IGIBD), Marco Cedola (SUMAI), Salvatore Leone (Direttore Generale AMICI Onlus), Massimo Medaglia (Regione Lombardia), Ambrogio Orlando (AO Ospedali Riuniti Villa Sofia, Palermo), Chiara Fattore (Ernst & Young), Federica Mammarella (AIFA)
Area clinica Dermatologia
Facilitatori:
Luca Bianchi (UOC Dermatologia, Policlinico Tor Vergata, Roma)
Partecipanti:
Anna Arcieri (SUMAI), Paolo Gisondi (Università degli Studi di Verona), Lorella Lombardozzi (Regione Lazio), Mara Maccarone (Presidente Associazione per la Difesa degli Psoriasici- ADIPSO), Luigi Naldi (AO Ospedali Riuniti di Bergamo), Chiara Vassallo (Ernst & Young), Sandra Petraglia (AIFA)
Area clinica Oncologia
Facilitatori:
Stefania Gori (Presidente Associazione Italiana di Oncologia Medica – AIOM)
Partecipanti:
Francesco De Lorenzo (Presidente Federazione Italiana delle Associazioni di Volontariato in Oncologia- FAVO), Rosaria Iardino (Fondazione The Bridge), Giovanni Poles (SUMAI), Fabrizio Nicolis (Presidente Fondazione AIOM), Alessandro Rambaldi (AO Papa Giovanni XXIII, Bergamo), Fausto Roila (Consigliere Collegio Italiano dei Primari Oncologi Medici Ospedalieri – CIPOMO), Giovanna Scroccaro (Regione Veneto), Edoardo Marco Di Cicco (Ernst & Young), Odoardo Maria Olimpieri (AIFA)
Tra i motivi di sottotrattamento, sono stati riportati la mancata applicazione delle linee guida, la carenza di percorsi per la presa in carico del paziente, la bassa affluenza dei pazienti ai centri prescrittori, la mancanza di reti territoriali adeguate. È interessante notare come spesso siano state indicate motivazioni legate soprattutto all’organizzazione aziendale e non solo ai costi dei farmaci.
Soprattutto dermatologi e gastroenterologi, ma anche reumatologi, hanno evidenziato come la percezione del farmaco biologico da parte del paziente giochi un ruolo molto importante nel trattamento e nella compliance, prima ancora di essere implicato in possibili switch: spesso i pazienti sono riluttanti ad intraprendere una terapia sistemica per curare una patologia apparentemente solo locale (ad esempio la psoriasi) oppure considerano il biologico come una terapia “sperimentale” o speciale, che incute timore e viene rifiutata. Da parte dei dermatologi è stata inoltre sollevata l’esigenza di poter disporre di un codice di esenzione come patologia cronica per la psoriasi di grado moderato/grave. I reumatologi hanno invece segnalato il problema dei lunghi tempi delle liste d’attesa, che si correla con il ridotto numero di specialisti.
Per quanto riguarda le soluzioni migliorative per il futuro, i suggerimenti si sono indirizzati verso la semplificazione dell’accesso alle terapie, con percorsi di presa in carico del paziente più efficienti, un maggiore coinvolgimento del paziente e del Medico di Medicina Generale, un migliore supporto alle farmacie territoriali.
Studi epidemiologici, registri di patologia e PDTA come strumenti per contrastare il gap informativo
Ai Tavoli hanno partecipato anche i rappresentati delle principali Associazioni di pazienti delle patologie trattate con biologici, che hanno fornito un contributo importante alla discussione sottolineando in particolare di avvertire forte il rischio di essere strumentalizzati, e di conseguenza di fare molto affidamento sui medici e sulle loro Società scientifiche per il ruolo di garante di una corretta informazione.
Infine, dai Tavoli tematici è emerso, come esigenza comune, l’auspicio ad attivare un dibattito condiviso sulla possibile destinazione delle risorse economiche recuperate grazie all’utilizzo dei farmaci biosiomilari e una costante collaborazione con le Regioni, tramite i flussi amministrativi e la pubblicazione di frequenti aggiornamenti sui dati raccolti.
Un auspicio accolto con entusiasmo dal Direttore Melazzini che, in conclusione di giornata, ha ribadito l’interesse di AIFA sul tema dei biosimilari, per le tante e complesse sfaccettature che presenta, e ha anticipato l’intenzione di mantenere in attività i Tavoli di discussione con incontri periodici ma anche di avviare una serie di progetti di ricerca indipendente condotti da AIFA per valutare la ricaduta dell’adozione dei biosimilari sul sistema, considerando anche i costi indiretti e sociali.


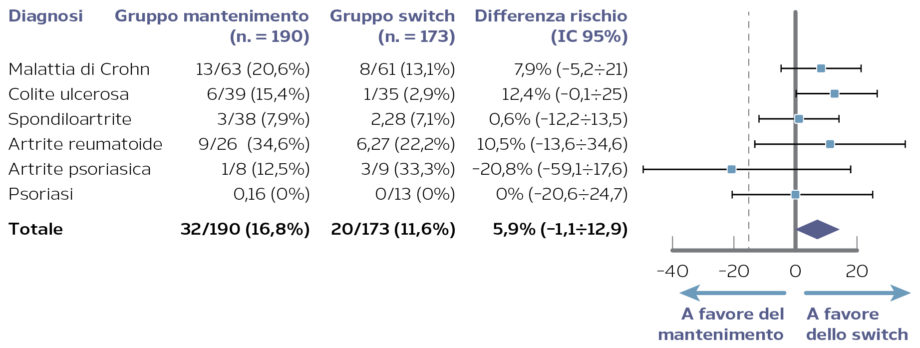



 Innanzitutto vorrei ricordare un breve antefatto: nel periodo in cui le Linee di indirizzo a carattere regionale non erano ancora state implementate e diffuse, i dermatologi del nostro Centro sono stati i primi a effettuare lo switch terapeutico dei pazienti in terapia con Remicade (Infliximab originator) a Infliximab biosimilare. La nostra scelta si è basata su due motivi fondamentali: innanzitutto la pari efficacia e sicurezza del biosimilare rispetto all’originator, e la conseguente consapevolezza di preservare la salute dei nostri pazienti; in secondo luogo, quello che riteniamo essere un obbligo morale del medico, e cioè garantire l’accesso alla cura a tutti i pazienti che ne hanno bisogno. Il costo importante delle terapie biologiche e l’aumento del numero di pazienti potenzialmente indirizzati a queste cure ci portano a considerare come opportuna la riduzione dell’entità della spesa, in modo da liberare i fondi per trattare più persone.
Innanzitutto vorrei ricordare un breve antefatto: nel periodo in cui le Linee di indirizzo a carattere regionale non erano ancora state implementate e diffuse, i dermatologi del nostro Centro sono stati i primi a effettuare lo switch terapeutico dei pazienti in terapia con Remicade (Infliximab originator) a Infliximab biosimilare. La nostra scelta si è basata su due motivi fondamentali: innanzitutto la pari efficacia e sicurezza del biosimilare rispetto all’originator, e la conseguente consapevolezza di preservare la salute dei nostri pazienti; in secondo luogo, quello che riteniamo essere un obbligo morale del medico, e cioè garantire l’accesso alla cura a tutti i pazienti che ne hanno bisogno. Il costo importante delle terapie biologiche e l’aumento del numero di pazienti potenzialmente indirizzati a queste cure ci portano a considerare come opportuna la riduzione dell’entità della spesa, in modo da liberare i fondi per trattare più persone.


 Recentemente il TAR del Piemonte ha pronunciato una sentenza a favore delle Linee di indirizzo emanate dalla Regione Piemonte sull’utilizzo dei biosimilari nelle patologie dermatologiche. Lei che ne pensa?
Recentemente il TAR del Piemonte ha pronunciato una sentenza a favore delle Linee di indirizzo emanate dalla Regione Piemonte sull’utilizzo dei biosimilari nelle patologie dermatologiche. Lei che ne pensa?



