La centralizzazione degli acquisti pubblici, in particolar modo in sanità, è ormai un dato di fatto. La definizione delle categorie merceologiche da acquistare obbligatoriamente attraverso iniziative dei soggetti aggregatori (vedi DPCM 24 Dicembre 2015 e DPCM 11 Luglio 2018) ha impresso un notevole impulso all’aggregazione della committenza. Le Regioni d’altro canto hanno visto negli acquisti un driver importante di efficientamento. L’assunto base della centralizzazione è che a una maggiore dimensione della gara, corrisponda necessariamente un maggior potere di acquisto e quindi la possibilità di ottenere condizioni più vantaggiose.
Prescindendo per un momento da considerazioni di merito sull’opportunità di orientare gli acquisti al prezzo, anziché al valore generato per il sistema e per il paziente, come dimostrano i risultati dell’analisi qui presentata, in realtà la relazione tra dimensione dell’acquisto e prezzo di aggiudicazione non è così immediata. A pesare, come si dirà in seguito, sono soprattutto le modalità con cui si costruiscono i lotti e il grado di competitività della procedura di selezione adottata.
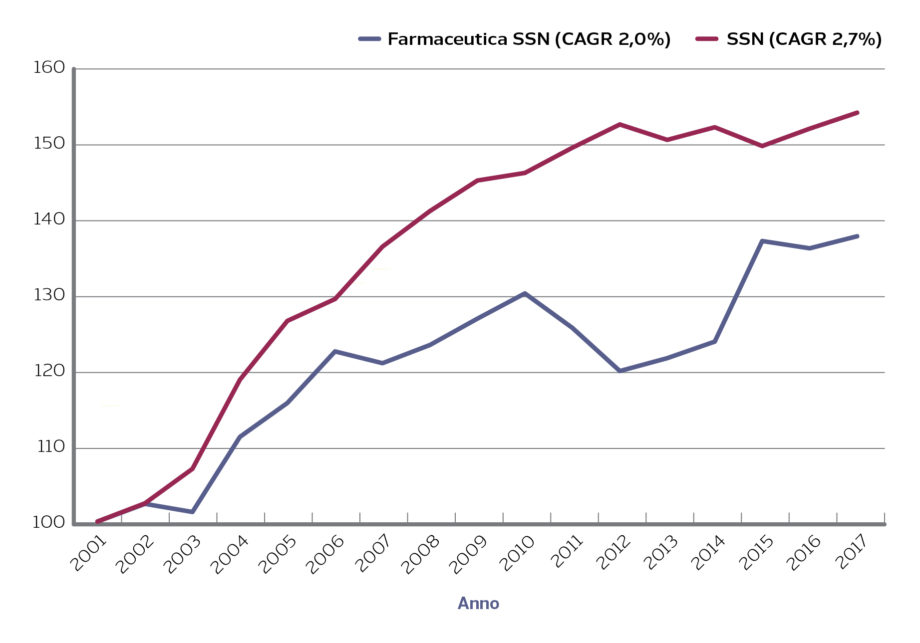
I farmaci sono stati, per il loro peso relativo sulla spesa (37% dell’acquisto di beni e servizi non sanitari nel 2018) e supposta facilità di aggregazione, al centro delle iniziative di centralizzazione. Dall’analisi dei dati disponibili sul portale dei soggetti aggregatori risulta, infatti, che i farmaci rappresentano circa il 50% delle iniziative chiuse, in corso e programmate dei soggetti aggregatori e il 63% in termini di valore stimato (Figura 1).
Figura 1. Numero e valore delle iniziative dei soggetti aggregatori
I soggetti aggregatori fanno (e faranno nel caso di iniziative programmate) ricorso a diversi strumenti (Tabella I) per l’acquisto di farmaci. Se in termini di numero di iniziative la convenzione è lo strumento più diffuso, i sistemi dinamici d’acquisto (SDA) hanno via via assunto maggiore importanza in termini di valore. Dai dati disponibili il valore delle convenzioni censite è pari a 36 miliardi di euro, quello dei SDA è pari a 39,6 a cui aggiungendo lo SDA Farmaci del 2017 di ARCA Lombardia non figurante nel database, si arriva a 45 miliardi di euro. La durata media delle iniziative è di circa 28 mesi, quella dei contratti di 31, per complessivi 5 anni. Gli accordi quadro (AQ) valgono 3,8 miliardi di euro. È bene ricordare che gli accordi quadro sono obbligatori per le gare di farmaci biotecnologici quando i prodotti in concorrenza sono più di tre, ovvero un originatore e almeno tre biosimilari.
Anno
ND
Altro*
AQ
Convenzione
Gara su delega
SDA
Totale
2014
20
2
22
2015
19
26
1
2
48
2016
48
2
112
12
4
178
2017
40
6
87
1
5
139
2018
9
28
14
171
4
1
227
2019
15
14
37
60
126
2020
7
4
4
12
27
2021
2
1
1
4
Totale
31
155
63
489
20
12
771
Tabella I. Strumenti utilizzati dai soggetti aggregatori
* Procedure negoziate e alcuni appalti specifici all’interno degli SDA (per cui di fatto si ha un double counting di alcune iniziative)
Il ricorso a strumenti diversi dalla classica convenzione e appalto ha un impatto sul mercato. Un sistema dinamico di acquisizione, infatti, consente, accorciando i tempi di pubblicazione, di tornare più frequentemente sul mercato lanciando confronti competitivi frequenti per l’aggiudicazione degli appalti specifici. Questo permette alle stazioni appaltanti di stringere contratti più brevi e approfittare dell’uscita di biosimilari e generici in tempi più rapidi. La conseguenza è potenzialmente quella di accrescere le pressioni competitive con conseguenti ricadute in termini di prezzi di aggiudicazione. Oltre agli strumenti, occorre inoltre considerare le modalità con cui vengono definiti i lotti, con un passaggio da lotti merceologici a lotti funzionali in cui poter mettere in competizione farmaci per cui è dimostrata l’equivalenza terapeutica.
Questi dati tuttavia ci mostrano soltanto gli input del processo, occorre, infatti domandarsi quali siano gli esiti, quindi gli impatti effettivi sui prezzi. A questo proposito uno studio recente (Armeni et al. OASI 2018) ha approfondito la performance del sistema di acquisto di specialità medicinali da parte dei soggetti pubblici del SSN, in termini di concorrenza generata nel mercato dei farmaci (numero di offerte per lotto, sconto aggiudicato rispetto al prezzo ex-factory, fattori che influenzano la non aggiudicazione). L’analisi ha considerato tutte le procedure di acquisto pubblicate dal 2006 al 2016 (fonte IHS) ed è stata strutturata al livello dell’ambito competitivo rilevante (tipicamente il singolo lotto o sub-lotto).
Dai risultati emerge, innanzitutto, una diffusa riduzione del numero di lotti per gara che potrebbe essere interpretata come lo specchio di una sempre più marcata aggregazione dei lotti e di acquisti di prodotti specialistici, il che spiega anche la crescente proporzione di lotti semplici. D’altro canto, questo potrebbe anche essere frutto del crescente ricorso nel tempo ai sistemi dinamici di acquisizione per cui a grandi gare multi-lotto si sostituiscono appalti specifici. Dallo studio, sorprendentemente, non emergono rilevanti risultati legati alla tipologia di lotto. Intuitivamente un lotto più aperto (unico) dovrebbe portare a più offerte. Questo è confermato solo in parte, dato che le gare per lotti unici hanno una probabilità di andare deserte minore rispetto a quelle per lotti semplici, ma solo se la gara è regionale o di area vasta, mentre nel caso di gare aziendali o di network, il lotto unico è associato a probabilità di non aggiudicazione maggiori.
In merito al fenomeno della mancata aggiudicazione lo studio osserva un numero crescente di lotti andati deserti. Questo potrebbe dipendere da requisiti di partecipazione sproporzionati, da basi d’asta inadeguate, ancora da condizioni di fornitura giudicate inappropriate dalle imprese. D’altro canto si potrebbe trattare di comportamenti deliberati da parte delle imprese con il risultato di generare proroghe di contratti in essere o ancora forzare le singole aziende a ricorrere a procedure negoziate con la motivazione di gara deserta. Questo fenomeno, oltre a comportare costi amministrativi non indifferenti, nei casi peggiori può condurre a indisponibilità di farmaci con una riduzione dell’accesso dei pazienti alle terapie farmacologiche.
Le analisi hanno rilevato una riduzione dell’aggiudicazione di gare da parte dei generici, indice di un sistema concorrenziale sempre più maturo, in cui anche i farmaci branded offrono condizioni di prezzo vantaggiose per il SSN. Tuttavia, in merito agli sconti in gara, emerge che la scontistica risulta più elevata quando la gara viene aggiudicata a generici o biosimilari. Aumentano in modo rilevante le aggiudicazioni di biosimilari, grazie alle prime scadenze di brevetti di molecole biotecnologiche.
Riguardo agli effetti della centralizzazione lo studio giunge ad alcune considerazioni interessanti. Se si guarda al livello di competizione, ovvero al numero di offerte ricevute per ogni lotto, si osserva una crescita del numero di offerte nel caso di procedure aperte e dinamiche di acquisto/accordi quadro. In particolare, nel caso di farmaci biotecnologici, un lotto aperto a originatore e biosimilare si traduce in un numero maggiore di offerte riducendo sensibilmente la probabilità che il lotto non venga aggiudicato, oltre che a generare uno sconto più consistente. Gli effetti totali però non sono univoci, a testimonianza di due dinamiche contrapposte: da una parte il maggiore effetto competitivo presunto sui prezzi può rappresentare un’importante barriera all’entrata, dall’altra non partecipare ad una gara regionale significa non cogliere rilevanti opportunità di mercato.
In merito alla dimensione del lotto e all’ente aggiudicatario, la ricerca ha mostrato effetti statisticamente significativi nonostante un’entità dell’effetto piuttosto contenuta. Non emerge chiaramente, infatti, che il processo di centralizzazione degli acquisti (ovvero le gare pubblicate dalle centrali) porti ad uno sconto maggiore. Nonostante le due limitazioni riportate nello studio e cioè: i) la valutazione della performance delle gare si è basata sugli effetti diretti (esito delle gare stesse) e non ha indagato le conseguenze indirette come ad esempio la disponibilità effettiva dei prodotti; e ii) i lotti non sono stati distinti tra lotti con una molecola e lotti con più molecole (equivalenza terapeutica), le evidenze emerse sono abbastanza chiare. Dai risultati della ricerca condotta da Armeni e colleghi risulta infatti che le procedure e la modalità di definizione dei lotti (per alcuni aspetti) sembrano pesare di più rispetto al livello di centralizzazione. In altre parole, il potere contrattuale non è il risultato della mera forza bruta, misurata in dimensione della gara, quanto della capacità delle stazioni appaltanti, centrali di committenza o singole aziende, di adottare una strategia di acquisto efficace.
A livello internazionale Toulemon (2018) osserva come l’appartenenza a un gruppo di acquisto comporti un impatto sui prezzi di acquisto dei medicinali, con una diminuzione media del 2% del prezzo di acquisto rispetto agli acquisti individuali. Occorre però distinguere tra tipologie di medicinali. Se da un lato non vi è alcun impatto significativo dell’acquisto di gruppo per i farmaci in esclusiva, per quanto riguarda i farmaci in cui vi è presenza sul mercato di biosimilari/generici, i prezzi medi sono inferiori del 9% quando gli ospedali fanno parte di un gruppo di acquisto rispetto a quando acquistano singolarmente.
Entrambi i risultati sembrano confermare che in questo caso la centralizzazione non alteri dinamiche di mercato già presenti, ma le possa rafforzare accrescendo i costi di non partecipazione alla gara perché un fornitore si troverebbe tagliato fuori non solo da un’azienda, ma da un’intera regione.
La domanda che resta per certi versi irrisolta è come si misuri l’efficacia di un acquisto. Riprendendo quanto detto in precedenza non è detto che il prezzo sia espressione del valore. I temi della disponibilità dei prodotti, dell’innovazione, dell’orientamento agli outcome, necessariamente devono essere presi in considerazione laddove si parli di acquisti che impattano direttamente sulla salute dei pazienti. La centralizzazione è nata principalmente per generare risparmi in un momento di forte stress finanziario dell’SSN. La capacità di risposta del sistema ai fabbisogni, tuttavia, richiederà in futuro di concentrarsi effettivamente sulla creazione di valore. Questo richiede siano prese scelte politiche e strategiche chiare su qual è il livello di cure che il sistema sanitario è disposto a offrire e quali risorse impegnare.
Il Medicinrådet, il consiglio danese che si occupa di valutare l’introduzione di nuovi farmaci ospedalieri e di verificare la consistenza dei benefici per i pazienti, ha valutato positivamente l’utilizzo del biosimilare di adalimumab in dermatologia, gastroenterologia e reumatologia, in tutte le indicazioni già autorizzate per il farmaco originator e per le seguenti categorie di pazienti:
pazienti che non sono mai stati in terapia con adalimumab;
pazienti già trattati in precedenza con adalimumab e che, per una recidiva di malattia, necessitano di riprendere il trattamento;
pazienti sottoposti a trattamento con adalimumab.
Inoltre, ha confermato l’assenza di motivazioni tecniche che giustifichino una limitazione del numero di switch tra i biosimilari di adalimumab, pur suggerendo che il numero di switch dovrebbe essere ridotto al minimo, anche per evitare un eventuale aumento di risorse correlate alla necessità di informare correttamente il paziente sulle diverse modalità di somministrazione e utilizzo dei farmaci. È sottolineata quindi l’importanza di supportare il paziente, al momento della decisione sullo switch, con informazioni scritte e orali, e si ricorda come sia l’agenzia del farmaco danese (Lægemiddelstyrelsen) sia l’EMA abbiano predisposto materiali ad hoc.
Nelle conclusioni del documento rilasciato dal Medicinrådet si affronta un ulteriore aspetto di interesse: il gruppo di lavoro interdisciplinare ha infatti stabilito che le differenze in termini di volume ed eccipienti riscontrabili nelle diverse formulazioni di adalimumab (originator e biosimilare) non ne compromettono la parificazione. Una base importante anche ai fini dell’approvvigionamento dei farmaci e della costituzione dei lotti di gara.
Secondo la definizione del Ministero della Salute italiano, l’Health Technology Assessment (HTA) è «un approccio multidimensionale e multidisciplinare per l’analisi delle implicazioni medico-cliniche, sociali, organizzative, economiche, etiche e legali di una tecnologia attraverso la valutazione di più dimensioni quali l’efficacia, la sicurezza, i costi, l’impatto sociale e organizzativo. L’obiettivo è quello di valutare gli effetti reali e/o potenziali della tecnologia, sia a priori che durante l’intero ciclo di vita, nonché le conseguenze che l’introduzione o l’esclusione di un intervento ha per il sistema sanitario, l’economia e la società».
L’HTA è nata nel 1976 negli Stati Uniti e in seguito si è diffusa in Europa, in America Latina e in Asia.
Lo sviluppo di molti nuovi farmaci negli ultimi anni è stato caratterizzato da un aumento di costi senza precedenti, che ha costretto i sistemi sanitari nazionali a ottimizzare ancora meglio le risorse a disposizione, nel tentativo di garantire il più possibile equità e universalità.
L’HTA completa e la Relative Effectiveness Assessment (REA) rapida, che valuta meno aspetti, sono entrate sempre più a far parte della procedura adottata dalle autorità nazionali per stabilire il prezzo e il rimborso delle tecnologie sanitarie. Vengono inoltre adottate dai policy maker per formulare politiche sanitarie che rispondano ai requisiti di sicurezza, efficacia, focus sul paziente e costo-efficacia.
La cooperazione sull’HTA a livello europeo
Le aziende che producono tecnologie sanitarie si trovano nella situazione di dover soddisfare i requisiti di ogni Paese, che variano notevolmente. Innanzitutto la costo-efficacia varia a seconda dei setting, così come il contesto politico e sanitario, le priorità nazionali e sociali. Inoltre i contenuti e la metodologia adottata per redigere l’HTA o la REA variano a seconda del sistema sanitario, dei processi di rimborso, della struttura socio-culturale e del PIL pro-capite.
Diversi Paesi europei hanno pertanto intrapreso un percorso per collaborare in merito all’HTA, al fine di incrementarne la qualità, l’efficienza e l’uso nei processi decisionali, ma sempre in accordo alle procedure e al quadro legislativo di ogni stato, secondo il motto “Evidence is global, decision is local”.
L’Unione Europea ha iniziato a dare impulso a progetti riguardanti la collaborazione sull’HTA a livello europeo nel 1993, arrivando a definirla una priorità nel 2004 e continuando tuttora a promuoverli e finanziarli.
Due enti sono responsabili di condurre la cooperazione sull’HTA:
l’HTA network, a cui partecipano tutti gli stati membri dell’Unione Europea, oltre a Islanda e Norvegia. Si tratta della “testa pensante” dell’organismo, che ha il compito di produrre policy paper e discutere sulle potenziali aree di collaborazione;
EUnetHTA Joint Action, a cui prendono parte Norvegia, Svizzera e tutti gli stati membri dell’UE ad eccezione del Lussemburgo. È il “braccio”, la componente scientifica e tecnica che esegue quanto deciso dall’HTA network.
Box. Articolo 15
Direttiva 2011/24/UE del Parlamento Europeo e del Consiglio del 9 marzo 2011 concernente l’applicazione dei diritti dei pazienti relativi all’assistenza sanitaria transfrontaliera
Sinteticamente, afferma che:
l’UE promuove lo scambio di informazioni tra stati relativamente all’HTA
i principi della HTA network sono: trasparenza, obiettività, indipendenza delle perizie, correttezza procedurale e opportune consultazioni degli stakeholder
uno degli obiettivi è evitare la duplicazione delle valutazioni
l’Unione Europea contribuisce a finanziare gli aspetti amministrativi e tecnici, lo sviluppo e la condivisione dei metodi di valutazione dell’HTA, la condivisione delle informazioni, anche da parte di tutti gli stakeholder, la comunicazione tra l’HTA network e l’UE
le misure intraprese dall’HTA network non devono interferire con i poteri decisionali dei singoli stati membri in tema sanitario
L’HTA network
L’HTA network prende le mosse dalla Decisione di Esecuzione della Commissione nel 2013 per dare attuazione all’articolo 15 della direttiva europea del 2011 concernente l’applicazione dei diritti dei pazienti relativi all’assistenza sanitaria transfrontaliera (Box).
Si tratta di una rete che connette gli enti che si occupano di HTA. Comprende anche un network di stakeholder (HTA Network Stakeholder Pool) in qualità di osservatori, tra cui pazienti, health provider (come Società medico-scientifiche), payer e industrie. Beneficia del supporto scientifico e tecnico dell’EUnetHTA Joint Action.
L’HTA Network ha il compito di fornire raccomandazioni sui meccanismi scientifici e tecnici di cooperazione.
In un reflection paper sui prodotti farmaceutici del 2016, suggeriva diverse aree di possibile collaborazione, successivamente sviluppate, almeno in parte, dalla EUnetHTA Joint Action 3:
fase premarketing:
early dialogues/consulenza scientifica tra aziende produttrici, enti regolatori e di HTA;
definizione e descrizione dell’applicazione di unmet medical need e innovazioni terapeutiche;
sviluppo di piani per aumentare i livelli di evidenza;
programmi di horizon scanning per identificare terapie emergenti con potenziale valore aggiunto;
promozione di ricerca e dialogo con gli stakeholder;
promozione della cooperazione sulle metodologie di HTA usate;
ingresso sul mercato:
condivisione delle informazioni tra enti regolatori e di HTA per identificare la popolazione eleggibile per un dato trattamento;
condivisione precoce delle informazioni tra enti regolatori e di HTA per accelerare l’accesso a nuovi farmaci da parte dei pazienti;
standardizzazione dei report di valutazione;
fase post-marketing:
stilare linee guida per il disegno degli studi post-marketing;
sviluppare dei late dialogues con l’azienda produttrice;
collaborazione sulla generazione di dati di real world;
altre aree di possibile cooperazione:
farmaci orfani;
medicina personalizzata;
vaccini;
coinvolgimento dei pazienti e del personale sanitario;
Patient Reported Outcome/qualità di vita;
esplorazione dei possibili canali di comunicazione tra EUnetHTA, enti che si occupano di HTA, autorità nazionali competenti ed EMA;
definizione delle modalità di partecipazione di tutti gli stakeholder nell’interazione tra enti regolatori e di HTA;
garanzia di trasparenza da parte di tutti gli stakeholder.
EUnetHTA
Su proposta della Commissione europea e del Consiglio dei ministri, nel 2005 è stato avviato il progetto European Network for Health Technology Assessment 1 (EUnetHTA 1). Esso connette organizzazioni nominate dai governi, agenzie regionali e organizzazioni no-profit che producono o contribuiscono all’HTA. Inizialmente costituita da 35 organizzazioni con un orizzonte temporale di azione previsto sino al 2008, si è in seguito espansa sino a comprendere 81 organizzazioni facenti parte di 29 diversi Paesi europei. Al primo progetto sono seguiti altri 4, come illustrato in tabella.
Nome progetto
Orizzonte temporale
Budget investito
Obiettivi
EUnetHTA Project
2006-2008
n.r.
Esaminare possibilità, vantaggi e svantaggi di una collaborazione tra più Paesi
EUnetHTA Collaboration
2009
n.r.
Implementare la collaborazione permanente dei Paesi europei sull’HTA ponendo le basi per una Joint Action
EUnetHTA JA1
2010-2012
6.000.000 €
Realizzare una cooperazione efficace e sostenibile sulla valutazione delle tecnologie sanitarie in Europa
EUnetHTA JA2
2012-2015
9.500.000 €
Rafforzare l’applicazione pratica mediante lo sviluppo di strumenti e approcci alla collaborazione tra stati europei sull’HTA
Definire meglio i modi per creare una struttura sostenibile per l’HTA in Europa
Sviluppare una strategia generale, corredata da proposte di principi e di maniere per l’implementazione, per una collaborazione europea sull’HTA sostenibile, secondo i requisiti definiti nell’articolo 15 della direttiva europea del 2011 concernente l’applicazione dei diritti dei pazienti relativi all’assistenza sanitaria transfrontaliera (Box)
EUnetHTA JA3
2016-2020
20.000.000 €
Sulla base dei risultati ottenuti nelle precedenti Joint Action, sviluppare una collaborazione europea volontaria e sostenibile fornendo informazioni sull’HTA in modo obiettivo, affidabile, tempestivo e confrontabile
Preparare il terreno agli sviluppi successivi al 2020
Storia di EUnetHTA
JA = Joint Action; n.r. = non riportato
EUnetHTA si basa sui principi di universalità delle cure, accesso alle cure di buona qualità, equità, solidarietà e sussidiarietà nell’Unione Europea. Intende inoltre raggiungere una buona efficienza nella produzione di HTA per poter garantire la sostenibilità ai sistemi sanitari, e usare come metodi la miglior evidenza, standard metodologici comuni, fiducia e trasparenza.
Si occupa, inoltre, di favorire la produzione e il corretto uso di HTA. EUnetHTA, fungendo da piattaforma indipendente basata sull’evidenza, consente alle agenzie di HTA di condividere informazioni e metodi sull’HTA, gestendo al contempo la comunicazione con tutti gli stakeholder. Sviluppa infine alleanze per rafforzare le evidenze alla base dell’HTA e usare le migliori competenze scientifiche disponibili.
Aggiornamenti: conferenza del 9 luglio 2018
Il 9 luglio 2018 si è tenuta a Bruxelles una conferenza sul tema “The way forward for HTA cooperation – the views of stakeholders”. Si sono riuniti più di 300 policy maker, healthcare provider, rappresentanti di associazioni di pazienti e altri esperti. In tale occasione gli stakeholder hanno confermato il loro supporto alla cooperazione europea nell’ambito dell’HTA, auspicandone una crescita a livello di strutturazione, sostenibilità ed efficienza. L’obiettivo è consentire ai pazienti di accedere il più in fretta possibile alle migliori cure disponibili, ma anche risparmiare, consentendo una maggior sostenibilità dei sistemi sanitari. Parallelamente si andrebbero anche a ottenere maggiori qualità, potere predittivo e trasparenza.
Sono state fornite raccomandazioni in merito alla necessità di:
coinvolgere pazienti e medici, per garantire maggiore trasparenza e maggior attenzione al paziente. Per aumentare l’utilità dei loro interventi, potrebbe essere utile investire nella loro formazione;
generare evidenze che tengano conto dei bisogni dei pazienti e dei decision maker dei sistemi sanitari, obiettivi raggiungibili mediante il mantenimento della trasparenza;
gestire l’incertezza della fase post-marketing, traendo le evidenze dai registri e dai dati di real world.
Il lavoro di tutti gli stakeholder sarà particolarmente importante anche per la fase che della cooperazione HTA europea che inizierà dal 2020.
Il rafforzamento della cooperazione a partire dal 2020
A gennaio 2018 la Commissione Europea ha presentato una proposta per rafforzare ulteriormente la cooperazione sull’HTA dopo il 2020 in merito a:
valutazioni cliniche congiunte dei farmaci (joint clinical assessment);
consultazioni scientifiche congiunte fornite dagli organismi di HTA alle aziende produttrici (joint scientific consultations o early dialogues o dialoghi precoci);
identificazione delle tecnologie sanitarie emergenti più promettenti (horizon scanning o esercizio di allerta precoce);
cooperazione volontaria su altri aspetti.
Rimarrebbero appannaggio dei singoli stati le valutazioni di aspetti non clinici (economici, sociali ed etici) e le attribuzioni di prezzi e rimborsi.
Nell’ottica della riduzione dei tempi e degli sprechi, i singoli stati non potrebbero più effettuare le valutazioni cliniche dei farmaci, ma dovrebbero basarsi unicamente sui report derivanti dai joint clinical assessment.
Gli stakeholder verrebbero coinvolti in tutte le forme di cooperazione sopra descritte e verrebbero riuniti in un network.
Queste proposte, benché ispirate da principi condivisi, non incontrano il favore di tutti, in quanto i vari stati presentano notevoli differenze nella scelta degli elementi da considerare nelle valutazioni cliniche (endpoint, ruolo degli stakeholder, qualità delle evidenze, metodi, ecc.). Inoltre, anche se gli aspetti clinici verrebbero considerati una volta sola a livello europeo, rimarrebbero comunque da eseguire le singole valutazioni degli aspetti non clinici e dunque non potrebbe essere garantita quella tempestività nell’accesso alle cure che ci si auspicava.
Tale proposta deve ancora essere vagliata dal Parlamento e dal Consiglio.
Bibliografia di riferimento
Ciani O, et al. Gli Stati membri alla prova della cooperazione in materia di valutazione delle tecnologie: la proposta Ue sull’HTA. Sanità 24, 9 luglio 2018
European Commission. Directorate-General for Health and Food Safety. Health systems, medical products and innovation. Medical products: quality, safety, innovation. HTA network reflection paper on “Synergies between regulatory and HTA issues on pharmaceuticals. Adopted by the HTA Network on 10 November 2016. Disponibile online su https://ec.europa.eu/health/sites/health/files/technology_assessment/docs/ev_20161110_co06_en.pdf (ultimo accesso settembre 2018)
European Network for Health Technology Assessment (EUnetHTA). Disponibile online su www.eunethta.eu (ultimo accesso settembre 2018)
Koçkaya G, et al. Pharmaceutical Market Access in Developed Markets. Torino: SEEd Medical Publishers, 2018
L 175/71. Decisione di esecuzione della Commissione del 26 giugno 2013 che stabilisce le norme per l’istituzione, la gestione e il funzionamento trasparente della rete di autorità nazionali o di organismi responsabili della valutazione delle tecnologie sanitarie (2013/329/UE). Gazzetta ufficiale dell’Unione Europea. Disponibile online su https://ec.europa.eu/health//sites/health/files/technology_assessment/docs/impl_dec_hta_network_en.pdf (ultimo accesso settembre 2018)
L 88/45. Direttiva 2011/24/UE del Parlamento Europeo e del Consiglio del 9 marzo 2011 concernente l’applicazione dei diritti dei pazienti relativi all’assistenza sanitaria transfrontaliera. Gazzetta ufficiale dell’Unione Europea. Disponibile online su https://eur-lex.europa.eu/legal-content/IT/TXT/PDF/?uri=CELEX:32011L0024&from=EN (ultimo accesso settembre 2018)
Procedura 2018/0018(COD). Proposta di Regolamento del Parlamento Europeo e del Consiglio relativo alla valutazione delle tecnologie sanitarie, che modifica la direttiva 2011/24/UE. Bruxelles: 31 Gennaio 2018. Disponibile online su https://eur-lex.europa.eu/legal-content/IT/TXT/HTML/?uri=CELEX:52018PC0051&from=EN (ultimo accesso settembre 2018)
La precisione terminologica è essenziale nel diritto farmaceutico
Solo negli ultimi anni è divenuto davvero indispensabile, per un giurista che intenda avvicinarsi ai temi delle equivalenze tra farmaci, comprendere esattamente il significato di molti concetti tecnici che non sono definiti dalla normativa e che appartengono più propriamente alla scienza farmaceutica e medica: biosimilarità, bioequivalenza, sovrapponibilità, comparabilità. Questa difficoltà terminologica è spesso impiegata nelle aule dei tribunali come tecnica ostruzionistica o quantomeno per rappresentare al giudice la complessità di una situazione che di per sé, al contrario, è di semplice approccio e soluzione.
È vero che non è sempre facile ricondurre la scienza farmaceutica a canoni e schemi prettamente giuridici: quando la diversità molecola è rilevante? Cosa è un principio attivo? Come valutare e verificare l’equivalenza tra due farmaci? Sono solo alcuni degli interrogativi la cui risposta potrà forse sembrare elementare per ogni farmacista ma che sono tutt’altro che semplici da affrontare per un giurista.
Le equivalenze tra farmaci – e si utilizza volutamente il termine al plurale, posto che come noto se ne può parlare sotto vari aspetti: farmacologico, chimico, terapeutico, eccetera – rappresentano chiaramente il settore dove maggiormente appare indispensabile utilizzare sinergicamente e correttamente nozioni farmaceutiche e criteri giuridici.
Spesso questa operazione è ostacolata dall’esistenza di due forze opposte.
Da un lato, infatti, vi è l’esigenza di assicurare nelle procedure di gara pubblica la più ampia concorrenza, sia per assicurare il buon funzionamento del mercato (dei farmaci) nel suo complesso, sia perché essa rappresenta il fattore principale attraverso il quale il sistema sanitario può ottenere ingenti risparmi senza diminuire il livello qualitativo delle prestazioni di assistenza farmaceutica.
Dall’altro, la comparazione concorrenziale in gara è possibile però soltanto quando le caratteristiche farmacologiche dei diversi farmaci permettano un sufficiente livello di similarità tra la composizione (equivalenza farmacologica), il dosaggio, la forma farmaceutica e la modalità di somministrazione (equivalenza d’uso) ovvero del risultato terapeutico, sia in termini di efficacia che di sicurezza (equivalenza terapeutica). Quando questo livello di similarità possa essere considerato sufficiente per autorizzare l’esercizio di comparazione è compito della scienza stabilire, sia, in generale, attraverso gli organi regolatori a ciò preposti sia, con riguardo al singolo paziente, attraverso le valutazioni del medico.
Semplificando, il duello è tra concorrenza e cosiddetta libertà prescrittiva.
Concorrenza vs libertà prescrittiva: l’articolo 68 del codice degli appalti non serve
Uno degli errori più ricorrenti è quello di applicare anche ai farmaci le norme e le categorie giuridiche genericamente previste dal codice degli appalti, senza effettuare alcuna operazione di interpretazione sistematica con le specificità del farmaco, e in particolare con le norme che ne disciplinano gli aspetti regolatori e le corrette modalità di comparazione. L’esempio più frequente di ciò sta nella applicazione tout court dell’art. 68 del codice degli appalti sulla equivalenza tecnica anche ai farmaci.
Questa operazione è senz’altro metodologicamente sbagliata, come lucidamente sottolinea la sentenza del T.A.R. per la Liguria, sez. II, n. 278 del 9 aprile 2018, secondo la quale le esigenze di tutela del paziente giustificano deroghe alla normale operatività del principio di equivalenza e dove, per tale ragione, sono previste norme specifiche. Dal punto di vista terminologico, quindi, non è possibile utilizzare per i farmaci il concetto di equivalenza funzionale né descrivere il prodotto da porre in gara attraverso la semplice aggiunta dell’espressione “o equivalente”.
Le norme che, nelle gare per l’acquisto di farmaci, sostituiscono l’art. 68 del codice degli appalti sono tre:
per i soli farmaci chimici, l’art. 7 del d.l. n. 347/2001 conv. in legge n. 405/2001, che afferma come due farmaci chimici possano essere considerati equivalenti solo se hanno lo stesso principio attivo, lo stesso dosaggio, forma farmaceutica e via di somministrazione;
per i soli farmaci biologici e biotecnologici, l’art. 15, comma 11 quater del d.l. n. 95/2012 conv. in legge n. 135/2012, che stabilisce come due farmaci aventi lo stesso principio attivo possano, e debbano, essere posti in concorrenza soltanto se vi è un rapporto di biosimilarità accertato da EMA o, più raramente, da AIFA (farmaci biosimilari);
per tutti i farmaci, chimici e biologici, il comma 11 ter della medesima disposizione, secondo il quale possono essere posti in concorrenza anche farmaci aventi principi attivi diversi purché vi sia tra essi un rapporto di equivalenza terapeutica accertato da AIFA. In questa ultima ipotesi, vi è solo da ricordare che, per i farmaci biologici, la concorrenza tra principi attivi diversi, pur se in equivalenza terapeutica, è esclusa dalla legge (è il già citato comma 11 quater a dirlo) qualora vi siano biosimilari di uno dei due originatori.
La procedura ibrida: un caso particolare di equivalenza?
L’interesse per la sentenza del T.A.R. per la Liguria sopra citata risiede principalmente nella soluzione offerta ad altro problema. In breve, il caso è il seguente: due farmaci di origine chimica sono stati posti in concorrenza dalla stazione appaltante in quanto aventi lo stesso principio attivo e perché ritenuti equivalenti, benché entrambi siano originatori. Il secondo farmaco, infatti, non è stato autorizzato (nella specie, da EMA) come farmaco c.d. generico bensì come ibrido, anche per la non trascurabile circostanza che il primo farmaco non aveva ancora perduto la copertura brevettuale. Il procedimento di autorizzazione, quindi, ha seguito la procedura di cui all’art. 10, comma 6, d.lgs. n. 219/06.
Questa particolare situazione non ricade nell’applicazione di alcuna delle tre disposizioni di legge elencate al paragrafo precedente: non nella prima ipotesi, perché i due farmaci non sono chimicamente identici pur avendo lo stesso principio attivo; non nella seconda, perché si trattava di farmaci di sintesi e non di farmaci biologici; e neppure nella terza, dato che l’equivalenza terapeutica è, di norma, una situazione che va accertata tra originatori aventi principio attivo differente, e in questo caso, e solo in questo caso, la legge richiede il necessario previo accertamento dell’equivalenza terapeutica da parte di AIFA.
La soluzione offerta dal giudice ligure muove da un presupposto di fondo inconfutabile: se la legge consente che siano posti in concorrenza in una gara d’appalto farmaci, siano essi chimici ovvero biologici, aventi principi attivi diversi purché le autorità regolatorie ne abbiano accertato l’equivalenza terapeutica tra (art. 15, comma 11 quater, d.l. n. 95/12 conv. in legge n. 135/12), allora a maggior ragione deve essere ritenuta legittima la concorrenza in gara tra farmaci chimici aventi lo stesso principio attivo, ancorché entrambi originatori, sempre a condizione che l’autorità regolatoria ne abbia accertato l’equivalenza terapeutica.
Nella particolare fattispecie esaminata dal T.A.R., in effetti tale accertamento era stato rinvenuto nel provvedimento autorizzativo da parte di EMA secondo il quale entrambi presentavano un profilo di qualità, sicurezza ed efficacia “comparabile”.
Se dal punto di vista prettamente logico la soluzione appare convincente, tuttavia essa non pare essere suscettibile di divenire una regola generale di equivalenza nelle gare d’appalto per l’acquisto di farmaci. Ciò perché non è sufficiente l’avere seguito la procedura ibrida per ottenere l’autorizzazione di un nuovo farmaco, ma occorre di volta in volta anche verificare se in detta procedura l’autorità regolatoria abbia espressamente dichiarato un rapporto di equivalenza (terapeutica) tra i due farmaci.
“Comparabile” non significa “equivalente”
Inoltre, l’esercizio di comparazione tra i due farmaci nel caso di procedura ibrida è ben diverso sia da quello che viene effettuato nel caso di procedura abbreviata abitualmente impiegata per i farmaci generici e per i farmaci biosimilari, sia da quello abitualmente compiuto per valutare l’equivalenza terapeutica tra due farmaci.
Va ricordato che il farmaco ibrido è quello che, pur avente lo stesso principio attivo rispetto a quello di riferimento, presenta non trascurabili differenze, perché, ad esempio, ha dosaggio o via di somministrazione diversi oppure un’indicazione leggermente differente dal medicinale di riferimento. Anche il farmaco biosimilare presenta differenze rispetto all’originatore ma la situazione è in questo caso ben altra: nel biosimilare, le differenze sono trascurabili in termini di qualità, di efficacia e di sicurezza del farmaco; nell’ibrido, invece, sono più marcate. Questa è la ragione per la quale il biosimilare si può giovare completamente degli studi e delle sperimentazioni relative al medicinale di riferimento, mentre nel caso del farmaco ibrido questi dati sono utilizzabili soltanto in parte, dovendo essere completati da studi suppletivi preclinici e clinici che diano evidenza della comparabilità.
Ciò dimostra che comparabilità ed equivalenza non sono la stessa cosa.
La “comparazione” di un farmaco ibrido a quello di riferimento è concetto giuridicamente e scientificamente differente sia da quello di “bioequivalenza” (utilizzato per i farmaci generici di sintesi e anche per i biosimilari) sia da quello di “equivalenza terapeutica” (utilizzato tra principi attivi diversi).
Certamente la “comparabilità”, che giustifica il ricorso alla procedura ibrida, è qualitativamente inferiore alla bioequivalenza, tanto che il farmaco è appunto ibrido, e non generico né biosimilare. Il legame tra i due farmaci è dunque forte nel caso del generico e del biosimilare, e invece debole nel caso dell’ibrido.
Ma anche l’equivalenza terapeutica sembra indicare un legame che, benché si attui tra principi attivi diversi, tuttavia appare più forte che non la semplice comparabilità: infatti, in entrambi i casi l’esame ha ad oggetto la valutazione dei profili di qualità, di sicurezza e di efficacia dei due farmaci, ma mentre il primo richiede che il risultato sia equivalente, il secondo si accontenta per così dire di un risultato anche solo “comparabile”.
Negli ultimi anni, i processi di acquisto di beni e servizi del sistema sanitario sono stati interessati da cambiamenti significativi, guidati prevalentemente dalla ricerca di soluzioni in grado di razionalizzare e contenere la dinamica crescente della spesa. Tali cambiamenti si inseriscono in un contesto in cui, sul piano finanziario, si registra un progressivo ridimensionamento delle risorse pubbliche, che si concretizza in stringenti vincoli per le aziende sanitarie, a fronte di un fabbisogno in continua crescita (si pensi, ad esempio, a fenomeni quali l’invecchiamento della popolazione e l’aumento delle patologie croniche). Si tratta di condizioni che, seppure di contesto e contingenti, hanno favorito la ricerca di soluzioni innovative di fatto attuate all’interno di due principali ambiti.
Il primo è legato all’inquadramento normativo degli appalti pubblici. I provvedimenti approvati nel corso degli ultimi anni, sia a livello comunitario che nazionale, sono caratterizzati dal riconoscimento di una maggiore flessibilità per le aziende sanitarie. L’introduzione di alcune procedure di selezione dei fornitori, quali il partenariato per l’innovazione, il dialogo competitivo, l’accordo quadro, rappresentano un importante sforzo del legislatore per andare incontro ad alcune esigenze specifiche dei professionisti aziendali responsabili della funzione acquisti.
Il secondo aspetto è invece collegato alle soluzioni istituzionali e gestionali adottate dalle aziende sanitarie:
il ripensamento del ruolo di Consip S.p.A., quale centrale d’acquisto nazionale con funzioni di stipula di convenzioni quadro per l’approvvigionamento di beni e servizi per la pubblica amministrazione, e più in generale come gestore del programma di approvvigionamento telematico;
il rafforzamento del ruolo dell’e-procurement, che oggi risulta complementare all’attività di Consip S.p.A., atteso che offre, rispetto a quest’ultima, un livello maggiore di flessibilità nei prodotti offerti e una più ampia attenzione alle specificità aziendali;
l’introduzione di logiche di accentramento degli acquisti in ottica inter-aziendale o regionale, tramite la costituzione di centrali d’acquisto, centrali di committenza, soggetti aggregatori o la stipula di convenzioni tra le aziende sanitarie.
A partire dagli inizi degli anni 2000 sono state intraprese numerose iniziative di centralizzazione e di coordinamento degli acquisti di beni e servizi in sanità, spesso, però, senza un unico approccio condiviso. Infatti, è solo negli ultimi anni che si è ritenuto di rafforzare il processo di razionalizzazione della spesa e di centralizzazione degli acquisti, con l’identificazione delle Centrali regionali di Committenza e la costituzione dei Soggetti Aggregatori (SS.AA.), di cui fanno parte, ex DL 24 aprile 2014, n.66, Consip S.p.A., una centrale di committenza per ciascuna regione italiana e altre figure (ad esempio città metropolitane e province) che abbiano fatto richiesta all’Autorità nazionale anticorruzione (ANAC) di iscrizione all’elenco Nazionale dei SS.AA. Attualmente i SS.AA. sono 32, secondo quanto deliberato da ANAC il 17 gennaio 2018 (Figura 1).
Figura 1. Elenco nazionale dei Soggetti Aggregatori (SS.AA.) previsto da delibera ANAC n. 31, 17 gennaio 2018
SUA = Stazione Unica Appaltante
I SS.AA. non sono sostitutivi delle centrali regionali di committenza già costituite; il loro compito è di indire le procedure di acquisto aggregate relative alle diverse categorie merceologiche di spesa; inoltre, hanno l’esclusività delle procedure di acquisto individuate dal Tavolo tecnico dei SS.AA., anch’esso istituito con DL 66/2014, cui è stato conferito il compito di definire entro il 31 dicembre di ogni anno la lista delle categorie di beni e servizi e delle relative soglie al superamento delle quali gli enti pubblici debbono acquistare attraverso i SS.AA.: a partire dal 2018, unitamente alle relative soglie di obbligatorietà, le categorie di beni e servizi sono state indicate dal DPCM 11 luglio 2018 (Tabella I).
Anche il Codice dei contratti è intervenuto in maniera decisa sul sistema di centralizzazione degli acquisti, sia per le aziende sanitarie che per gli enti locali. Innanzitutto, ha previsto forme e modalità differenti di acquisto a seconda del valore della fornitura (Tabella II):
al di sotto del valore di 40.000€, le stazioni appaltanti possono effettuare, direttamente e autonomamente, acquisti di forniture e servizi, anche attraverso ordini sugli strumenti messi a disposizione dalle Centrali di Committenza (art. 37 c. 1);
al di sopra di questo importo e fino al valore della soglia comunitaria (pari a 221.000€), è stato introdotto il sistema della qualificazione, a mente del quale le stazioni appaltanti in possesso dei requisiti di cui all’art. 38 possono effettuare direttamente acquisti attraverso gli strumenti telematici di negoziazione messi a disposizione dalle Centrali di Committenza (convenzioni, accordi quadro, mercato elettronico). In caso di indisponibilità di tali strumenti, anche in relazione a singole categorie merceologiche, le stazioni appaltanti possono ricorrere alle centrali di committenza, o a forme di aggregazione, oppure possono svolgere una procedura ordinaria. Se invece le stazioni appaltanti non sono in possesso dei requisiti di qualificazione, devono necessariamente ricorrere ad una Centrale di Committenza, oppure devono aggregarsi con una o più stazioni appaltanti aventi la necessaria qualifica (art. 37, c. 3);
sopra la soglia comunitaria, le stazioni appaltanti devono comunque obbligatoriamente ricorrere alle Centrali di Committenza; in assenza di convenzioni o gare attive, possono procedere attraverso lo svolgimento di una procedura ordinaria. Naturalmente, come visto in precedenza, in relazione alle categorie merceologiche contenute nel DPCM 24/12/2015 (e futuri DPCM, con cadenza biennale) permane un obbligo assoluto di adesione, anche rispetto a convenzioni sottoscritte da altre regioni (con unico limite del rispetto del plafond contrattuale).
Soglia di rilevanza comunitaria per i contratti pubblici di forniture e di servizi aggiudicati dalle amministrazioni diverse dalle autorità governative centrali
Farmaci, vaccini, ausili per incontinenza (ospedalieri e territoriali), medicazioni generali, aghi e siringhe, servizi integrati per la gestione delle apparecchiature elettromedicali, servizi di pulizia/ ristorazione/lavanderia per gli enti del Servizio Sanitario Nazionale, servizi di smaltimento rifiuti sanitari, vigilanza armata, guardiania, guanti (chirurgici e non), suture
40.000 €
Tabella I. Categorie di beni e servizi da centralizzare e relative soglie
Importo economico (Iva esclusa)
Modalità di acquisto
Sotto 40.000€ (art. 37, c. 1)
Acquisti diretti e autonomi
Da 40.000€ a soglia comunitaria (221.000€)
Se la stazione appaltante è in possesso della necessaria qualificazione, utilizzo autonomo degli strumenti telematici di negoziazione messi a disposizione delle Centrali di Committenza
In caso di indisponibilità di tali strumenti, ricorso a procedura ordinaria (art. 37, c. 2)
Se la stazione appaltante non è in possesso della necessaria qualificazione, ricorso a Centrale di Committenza o Aggregazione di una o più stazione appaltanti aventi la necessaria qualifica (art. 37, c. 3)
Sopra soglia comunitaria (221.000€)
Utilizzo autonomo degli strumenti telematici di negoziazione messi a disposizione delle Centrali di Committenza
Ricorso a Centrale di Committenza
Ricorso a procedura ordinaria
Tabella II. Modalità di acquisto previste dal Codice (D. Lgs. 50/2016) secondo le soglie di riferimento
Per le aziende sanitarie, le norme del Codice dei contratti vanno integrate con i provvedimenti specifici in materia di spesa, già richiamati in precedenza, che hanno disciplinato ambiti e modalità di acquisto nel settore sanitario e che sottolineano gli obblighi di ricorso a Consip S.p.A. o alle Centrali di acquisto regionali, per i beni e servizi stabiliti con provvedimenti governativi. Il Codice dei contratti ribadisce che le Centrali di Committenza possono agire sia come centrale di acquisto (ovvero aggiudicare appalti, stipulare ed eseguire i contratti per conto delle amministrazioni aggiudicatrici e degli enti aggiudicatori), sia come gestore di procedure di gara (ovvero stipulare accordi quadro ai quali le stazioni appaltanti qualificate possono ricorrere per l’aggiudicazione dei propri appalti; oppure gestire sistemi dinamici di acquisizione e mercati elettronici). Il Codice conferma, infine, il ruolo di Consip S.p.A. quale Centrale di Acquisto, anche attraverso le convenzioni quadro di cui all’articolo 26 della legge 23 dicembre 1999, n. 488.
In sintesi, quindi, per le aziende del Servizio Sanitario Regionale, in relazione agli acquisti delle categorie di prodotti riportate nel DPCM (tra cui i farmaci), esiste in prima battuta l’obbligo di ricorso alle convenzioni della Centrale regionale di riferimento o, in mancanza, di Consip. In assenza sia di convenzioni attive con la Centrale regionale, sia con Consip, le aziende sono obbligate a ricorrere in via esclusiva alle Centrali di Committenza iscritte nell’elenco dei Soggetti Aggregatori, come individuate dalla Centrale regionale di committenza di riferimento. In assenza, permane l’obbligo di ricorso a strumenti di acquisto e negoziazione telematici messi a disposizione da Consip o dalla Centrale regionale di riferimento.
Centrali regionali di Committenza e Soggetti Aggregatori rafforzano il processo di razionalizzazione della spesa e centralizzazione degli acquisti
Pertanto, in presenza di una convenzione attiva con la Centrale di riferimento, le aziende del Servizio Sanitario non sono obbligate ad aderire alla nuova gara Consip; sarebbero obbligate solo in assenza di convenzione o gare attive nella propria centrale di riferimento.
A valle delle procedure utilizzate, le amministrazioni spesso fanno ricorso a differenti strumenti di acquisto: l’accordo quadro e la convenzione quadro, stipulate, ai sensi della normativa vigente, da Consip e dai Soggetti Aggregatori.
È nel contesto appena disegnato che si inserisce la gara di farmaci biologici effettuata da Consip, con bando pubblicato in data 23/11/2017.
Come richiamato in precedenza, Consip generalmente opera attraverso convenzioni, o accordi quadro. La convenzione consiste in un contratto quadro, con cui Consip o le Centrali di Committenza regionali individuano – a seguito di procedura di gara – uno o più fornitori, che si impegnano ad accettare ordini dalle amministrazioni di riferimento fino al raggiungimento del massimale contrattuale. Il modello di funzionamento si basa su un rapporto trilaterale, reso disponibile dalla Centrale di Committenza e che si perfeziona direttamente tra fornitore e amministrazione richiedente. Le convenzioni ex art. 26 della legge 23 dicembre 1999, n. 488 sono idonee per approvvigionamenti standardizzabili e, attraverso l’aggregazione della domanda, consentono di ottenere rilevanti economie di scala sia in termini di processo sia di risparmio sugli acquisti. I fornitori aggiudicatari di una gara si impegnano ad accettare le condizioni di prezzo, durata e oggetto della fornitura stabilito dalla Convenzione.
L’accordo quadro, disciplinato dall’art. 54 del Codice dei Contratti, consiste in un accordo concluso tra una o più amministrazioni aggiudicatrici e uno o più operatori economici, il cui scopo è stabilire le clausole relative agli appalti da aggiudicare durante un dato periodo, in particolare per quanto riguarda i prezzi e le quantità previste. La durata dell’accordo quadro non può superare i quattro anni (salvo casi eccezionali che richiedono obbligatoriamente una motivazione da parte della stazione appaltante), al fine di evitare meccanismi distorsivi della concorrenza.
La scelta del contraente attraverso lo strumento dell’accordo quadro si articola in due fasi. Nella prima, l’amministrazione provvede all’indizione della gara, secondo le procedure ordinarie previste dalla normativa (aperta, ristretta, negoziata). Tale fase si conclude con l’individuazione degli operatori economici idonei e la sottoscrizione del contratto. La seconda fase prevede una scelta, tra i diversi fornitori aggiudicatari, per l’assegnazione dei contratti relativi ai singoli ordinativi, al termine del quale l’amministrazione aggiudicatrice e i fornitori concludono i singoli contratti applicativi dell’accordo.
La gara Consip sui biologici ha permesso un risparmio medio del 18%
L’utilità del ricorso all’accordo quadro appare elevata per alcune tipologie di beni e servizi, quali quelli ad alto contenuto tecnologico, e in caso di acquisti ripetitivi e omogenei, che vengono in questo modo unificati in una sola procedura di gara ad evidenza pubblica, con un consequenziale probabile vantaggio economico sia in termini di riduzione dei prezzi d’acquisto che in termini di riduzione delle spese amministrative derivanti dall’indizione della gara. Infine, l’accordo quadro manifesta la propria utilità per l’acquisto di quella particolare categoria di beni soggetta ad un elevato grado di “personalizzazione” rispetto all’utilizzatore del bene stesso. In campo sanitario, si fa riferimento a quei beni per i quali la manualità e l’esperienza da parte dei clinici sembrano essere fattori fondamentali rispetto al buon esito delle prestazioni – si pensi ad esempio ad alcuni tipi di dispositivi medici (quali le suture chirurgiche). In questo caso, l’accordo quadro rappresenta il punto di incontro fra l’esigenza, di natura prevalentemente amministrativa, di ridurre i costi di approvvigionamento (che deriverebbero sia dai minori costi amministrativi che dalla riduzione sui prezzi d’acquisto in seguito al confronto competitivo fra le parti dell’accordo) e la necessità, di natura prevalentemente clinica, di sviluppare e mantenere nel tempo quella “confidenza” con gli strumenti di lavoro, elemento fondamentale, quasi imprescindibile, per garantire la qualità delle prestazioni erogate.
Lotto
Principio attivo
Aggiudicatario
1
Filgrastim
Pfizer Italia S.r.l.
Sandoz S.p.A.
2
Infliximab
Biogen Italia S.r.l.
Mundipharma Pharmaceuticals
3
Follitropina alfa
Teva Italia S.r.l.
Gedeon Richter S.p.A.
4
Etanercept
Sandoz S.p.A.
Biogen Italia S.r.l.
5
Insulina glargine
Nessuna offerta
6
Rituximab
Mundipharma Pharmaceuticals
Sandoz S.p.A.
7
Epoetina
Pfizer Italia S.r.l.
Sandoz S.p.A.
Tabella III. Gara Consip biologici: aggiudicatari dei singoli lotti
Come prevede la norma, l’accordo quadro può sancire due differenti modalità operative:
l’individuazione dell’operatore economico (tra quelli selezionati e con cui è concluso l’accordo) che effettuerà la prestazione avviene sulla base di decisione motivata, in relazione alle specifiche esigenze dell’amministrazione (in questo caso, l’accordo contiene tutti i termini e le condizioni che disciplinano le prestazioni dei servizi e forniture);
l’individuazione dell’operatore economico che effettuerà la prestazione avviene attraverso la riapertura del confronto competitivo (questo può avvenire sicuramente se l’accordo quadro non contiene tutti i termini e le condizioni che disciplinano le prestazioni dei servizi e forniture, ma anche su precisa scelta dell’amministrazione, e dichiarata nella documentazione di gara).
La modalità di aggiudicazione attraverso scelta motivata permette sicuramente di avere tempi più rapidi e un maggior coinvolgimento di tutti gli operatori.
In conformità alle sue facoltà discrezionali, Consip ha effettuato, mediante una procedura aperta, un accordo quadro, della durata di 12 mesi, aventi le caratteristiche di seguito riportate. L’importo massimo stimato per la gara era di € 460.000.000, suddiviso in 7 lotti merceologici:
lotto 1: filgrastim (€ 48.000.000);
lotto 2: infliximab (€ 30.000.000);
lotto 3: follitropina alfa (€ 18.000.000);
lotto 4: etanercept (€ 126.000.000);
lotto 5: insulina glargine (€ 55.000.000);
lotto 6: rituximab (€ 63.000.000);
lotto 7: epoetina (€ 120.000.000).
Ogni lotto prevede un dosaggio minimo per consentire la partecipazione dei fornitori.
La gara è stata aggiudicata sulla base del criterio del prezzo più basso, in quanto i prodotti presentano caratteristiche standardizzate (ossia le caratteristiche sono già definite dal produttore e non sono modificabili su richiesta della stazione appaltante, ovvero sono rispondenti a determinate norme nazionali, europee o internazionali).
La procedura centralizzata di acquisto dovrebbe puntare all’ottimizzazione del rapporto costo-efficacia
La gara, aggiudicata a giugno 2018, per 6 lotti su 7 (in quanto per uno non sono state presentate offerte) ha registrato ribassi che variano dal 14 al 48%, consentendo un risparmio medio stimato, rispetto ai prezzi praticati oggi alle strutture sanitarie pubbliche, pari al 18%, per un valore di circa 140 milioni di Euro sul totale della spesa (Fonte: Consip S.p.A.). Aggiudicata a tutti i concorrenti in gara, consentirà ai clinici un’ampia flessibilità nella scelta dei farmaci più idonei alle esigenze cliniche del paziente. Infatti, l’accordo quadro prevede che la seconda fase (definizione dell’appalto specifico) non avvenga attraverso un secondo confronto competitivo, bensì attraverso decisione motivata in relazione alle specifiche esigenze (Tabella III).
L’analisi della gara Consip sui farmaci permette di effettuare alcune considerazioni tenuto conto del carattere profondamente innovativo della gara.
Innanzitutto, la centralizzazione dei processi di acquisto, pur essendo un fenomeno ormai acquisito e difficilmente reversibile, richiede ancora un affinamento nei modelli di applicazione (accanto a soluzioni più evolute, costruite con il forte coinvolgimento degli attori in gioco, si trovano modelli ancora embrionali e semplicistici); risulta necessario completare la realizzazione di un sistema di acquisti, in cui Consip S.p.A. operi a stretto contatto e in coordinamento con le Centrali di committenza regionali e i Soggetti Aggregatori, anche per evitare facili duplicazioni e sovrapposizioni.
In secondo luogo, gli obiettivi dei processo di centralizzazione non dovrebbero consistere solo nel perseguimento della riduzione del costo per input, ma soprattutto nell’ottimizzazione del rapporto costo-efficacia, tenendo in considerazione l’intera catena di offerta (ricerca, sperimentazione, produzione, distribuzione, logistica, assistenza, ecc.) e tutte le ricadute della decisione di acquisto (governo clinico, compliance del paziente, tempi di attraversamento dei processi, ecc.).
Risulta fondamentale, infine, accompagnare i processi di centralizzazione ad una maggior implementazione degli strumenti di natura aziendale: lo sviluppo di politiche di marketing d’acquisto (studio sistematico dell’ambiente, dei mercati, dei prodotti, dei fornitori); il miglioramento delle informazioni gestionali (lo sfruttamento del potere negoziale; la ricerca di diminuzione dei costi di transazione; una maggiore attività di benchmarking sui prezzi per le tipologie di acquisto); la gestione proattiva dei fornitori (snellimento dei processi; coinvolgimento anticipato dei fornitori; ricerca di comunicazioni più chiare) sono elementi critici per le attività di programmazione degli acquisti che rappresentano la base della corretta impostazione di una gara.
La gestione della spesa farmaceutica è una voce fondamentale nei bilanci delle Regioni, non solo per il suo peso economico ma anche per la complessità dell’equilibrio tra le risorse disponibili e la domanda di salute. Quali sono state le opportunità e le criticità che le Regioni hanno dovuto affrontare nel governo del fondo per i farmaci innovativi?
Abbiamo qui riunito le voci di diversi stakeholders, che possono offrirci il punto di vista di alcune delle professionalità coinvolte nel governo della spesa regionale.
Il commento di Giovanna Scroccaro
Direttore, Direzione Farmaceutico-Protesica-Dispositivi Medici dell’Area sanità e Sociale della Regione Veneto
Come viene gestito il fondo farmaci innovativi nella sua Regione e come viene ripartito tra le diverse aziende sanitarie?
Va innanzitutto chiarito che il Ministero della Salute concorre a rimborsare le Regioni a statuto ordinario e la Sicilia in proporzione alla spesa sostenuta dalle Regioni medesime, così come indicato nella legge finanziaria n.232/2016. Qualora la spesa registrata a livello nazionale ecceda il valore fissato per i due fondi pari a 500 milioni ciascuno, la quota di spesa non coperta dai rispettivi fondi resta a carico delle singole Regioni.
È questo il motivo per cui la Regione del Veneto anche sui fondi dei farmaci innovativi effettua una programmazione della spesa e attribuisce alle aziende sanitarie un finanziamento parametrato sulla casistica attesa. L’attribuzione dei finanziamenti viene aggiornata a metà anno sulla base del numero dei pazienti effettivamente trattati nei centri autorizzati e/ o di nuove intervenute esigenze. La ripartizione dei fondi riguarda sia i farmaci innovativi oncologici che quelli non oncologici.
A fine anno, la Regione provvede a ripartire il finanziamento alle strutture sanitarie secondo quanto rilevato dai flussi di consumo e dai Registri AIFA. L’analisi dei registri AIFA è fondamentale quando un farmaco è autorizzato in commercio per più di una indicazione terapeutica, ma il requisito di innovatività, e quindi l’accesso al fondo, non è esteso a tutte le indicazioni, perché solo il registro – e non i consumi – può fornire le informazioni dettagliate sulle indicazioni di utilizzo.
Nella Regione Veneto, come avviene l’erogazione del fondo alle strutture private accreditate?
Nella nostra Regione l’uso dei farmaci innovativi non è molto consistente nelle strutture private accreditate; per tale motivo ci siamo organizzati facendo acquistare i farmaci necessari dalla Azienda Sanitaria in cui insiste la struttura privata, così come previsto dal Decreto ministeriale del 16 febbraio 2018. Infatti, il decreto prevede che la spesa regionale di competenza per singola Regione sia calcolata dalle dispensazioni rilevate attraverso i registri di monitoraggio AIFA sia per le strutture pubbliche che private, purché per queste ultime l’acquisto sia stato effettuato dall’ente del SSR di natura pubblica.
Come funziona il fondo per i pazienti che vengono trattati al di fuori della propria Regione di residenza?
Così come prescritto dalla normativa, in Regione Veneto abbiamo in cura – anche se con casistica limitata – pazienti che sono residenti in altre Regioni. Non abbiamo registrato nessun problema di gestione o capienza del fondo. Sappiamo peraltro che in altre Regioni la distribuzione non viene sempre garantita. Va inoltre ricordato che le norme prevedono che, al momento della redistribuzione a livello nazionale della spesa per i farmaci innovativi, in caso di superamento del tetto massimo del fondo, il rimborso per i pazienti fuori Regione venga garantito, mentre rimane a carico delle singole Regioni la quota spesa per i propri residenti.
Il fondo farmaci innovativi viene finanziato a livello nazionale, ma l’erogazione e il monitoraggio dei farmaci avviene a livello regionale: nella sua esperienza, come si armonizzano questi due piani? Ci sono elementi di criticità?
Il problema principale è rappresentato dal fatto che le Regioni non hanno a disposizione un aggiornamento mensile aggiornato sui consumi nazionali del fondo. La richiesta di una maggiore tempestività informativa sulla capienza del fondo è stata avanzata dalle Regioni in diverse occasioni perché riveste un’importanza fondamentale: poiché la gestione del fondo è a livello nazionale, la singola Regione, nel corso dell’anno, può monitorare il proprio consumo, ma non può sapere in tempo reale né quanto stanno spendendo le altre Regioni né se il tetto di spesa è stato raggiunto. È vero che l’AIFA pubblica con cadenza mensile il report del monitoraggio della spesa farmaceutica nazionale e regionale ma i report vengono pubblicati anche parecchi mesi dopo il periodo di riferimento.
L’informazione completa viene resa nota solo alla fine dell’anno, al momento del consuntivo; se le matrici di consumo nazionale aggiornate venissero fornite mensilmente alle Regioni, la situazione sarebbe più facilmente gestibile.
Se il fondo per i farmaci innovativi venisse gestito diversamente, ripartendolo in misura proporzionale alla quota di accesso al fabbisogno sanitario standard di ciascun anno di riferimento, certamente la gestione dei fondi e la programmazione regionale sarebbero semplificate: la Regione Veneto saprebbe già ad inizio anno che la sua quota del fondo innovativi è pari, ad esempio, all’8% del fondo complessivo e potrebbe organizzare la ridistribuzione tra le strutture sanitarie. Questa modalità di ripartizione ben si presta ai farmaci ove non esistono concrete dimostrazioni di un differente fabbisogno regionale perché correlato a variabilità epidemiologica. Certamente nel caso in cui la prevalenza di malattia sia diversa tra le Regioni, una ripartizione collegata alla quota di accesso potrebbe risultare poco equa. In ogni caso la proposta di attribuire i fondi in base alla quota di accesso non è percorribile perché la Legge che ha istituito i fondi innovativi prevede specificatamente che le Regioni vengano rimborsate in base all’effettivo consumo.
Rimborsare sulla base del consumo può peraltro incentivare scelte poco virtuose, quali l’impiego dei farmaci innovativi (nel caso degli oncologici) anche quando potrebbero essere utilizzati farmaci meno costosi, ma non innovativi, la cui spesa è quindi completamente a carico delle Regioni.
Non solo: in caso di superamento del fondo nazionale anche le Regioni che avessero usato con molta oculatezza i farmaci innovativi, potrebbero non vedersi garantito il completo rimborso di tutta la spesa sostenuta.
Da un punto di vista più generale, al di là del meccanismo e delle procedure, io credo che la norma meriterebbe di essere rivista perché può creare un incentivo al consumo, anche a scapito dell’appropriatezza. Certo, l’appropriatezza sul singolo farmaco rimane garantita dai Registri AIFA però, in un panorama di diverse terapie disponibili, i clinici possono adottare strategie diverse: ad esempio, c’è chi opta direttamente verso l’utilizzo del farmaco innovativo ma c’è anche chi può ritenere ancora valida un’altra alternativa, prima di passare al farmaco che accede al fondo. L’attuale sistema di rimborso può creare una “corsa” competitiva all’uso dei fondi poco virtuosa e non necessariamente correlata alla epidemiologia e ai fabbisogni. A mio modo di vedere, salvo alcuni farmaci per i quali ci sia una concreta dimostrazione delle differenze epidemiologiche tra Regioni, sarebbe preferibile ripartire il fondo allo stesso modo con cui viene ripartito il Fondo Sanitario Nazionale, demandando alle Regioni il compito di rispettare i Livelli essenziali di assistenza e nel caso specifico di garantire l’accesso ai farmaci innovativi a tutti coloro che ne hanno bisogno. Rimarrebbe in capo al livello nazionale il compito di mettere in atto tutte le verifiche necessarie a garantire il rispetto degli adempimenti.
Il commento di Anna Maria Marata
Coordinatore della Commissione Regionale del Farmaco, Regione Emilia Romagna. Direzione generale Cura della persona, Salute e Welfare
Come viene gestito l’accesso al fondo farmaci innovativi nella sua Regione e la ripartizione tra le diverse aziende sanitarie?
In Emilia Romagna la gestione dell’accesso al fondo per i farmaci innovativi dipende dalla classe di farmaci, con percorsi differenti per i farmaci oncologici e non oncologici. Il nostro punto di partenza è sempre la gestione clinica ottimale dei pazienti, con l’individuazione della casistica da trattare e il confronto con gli stakeholder coinvolti (medici, farmacisti, amministratori, rappresentanti dei pazienti) per far emergere le problematiche cliniche e/o organizzative legate alla singola patologia o al singolo farmaco. Su questa base si inserisce il governo della spesa che deve seguire direttrici diverse a seconda delle peculiarità evidenziate nel percorso precedente.
La gestione dei farmaci innovativi per l’epatite C, che è stata la nostra prima “palestra” di governo su questo tema a partire dal 2015, ha coinvolto migliaia di pazienti, con una serie di farmaci entrati in commercio in un periodo molto breve e che prevedono un trattamento limitato nel tempo (al massimo 24 settimane). Il nodo centrale in questo caso era ed è l’individuazione dei pazienti e la loro gestione ottimale applicando i criteri stabiliti a livello centrale e non discutibili, ma cercando di ottimizzare le risorse disponibili al fine di trattare il maggior numero di pazienti possibili con le risorse economiche messe a disposizione dal fondo. Nel 2015 (primo anno di disponibilità di tali farmaci) nella nostra Regione c’è stato un ampliamento regionale rispetto alla quota messa a disposizione dal fondo nazionale.
Una situazione ben diversa è stata quella che abbiamo affrontato con ivacaftor per la fibrosi cistica oppure con nusinersen per l’atrofia muscolare spinale (SMA): in quest’ultimo caso, il numero di pazienti da trattare è molto ristretto, ma il problema principale da affrontare riguardava il livello logistico/organizzativo, considerato che per la prima volta si doveva predisporre una somministrazione cronica per via intratecale, che rappresenta una modalità piuttosto complicata in quanto i pazienti devono sottoporsi ad una puntura spinale per ogni trattamento. Insieme ai prescrittori abbiamo dovuto individuare, come sempre, il numero di pazienti candidati al trattamento per ipotizzare l’impatto economico della nuova terapia ma anche, e questo è un elemento peculiare di nusinersen, come organizzare la somministrazione. La soluzione elaborata è stata quella di individuare un numero ristretto di centri prescrittori affiancati da strutture che possano somministrare il farmaco e supportare i centri prescrittori per raggiungere tutti i pazienti da trattare. Abbiamo poi dovuto condividere fra i prescrittori/somministratori una procedura per rendere ottimale e omogenea la somministrazione.
Il nostro approccio è sempre quello di gestire insieme ai nostri clinici la strategia più opportuna per far sì che si ottenga il massimo dell’efficienza avendo il controllo e il governo di quello che sta succedendo anche a livello economico. Il metodo è sempre quello di attivare dei gruppi di lavoro regionali cui delegare la produzione di documenti (raccomandazioni terapeutiche, linee guida, linee di indirizzo) che consentano un uso appropriato ed efficiente dei farmaci. Ogni paziente dovrà avere il farmaco più appropriato per gestire la sua patologia, ma il clinico nella scelta terapeutica, a parità di efficacia e sicurezza, dovrà considerare anche il costo dei farmaci al fine di perseguire l’uso ottimale delle risorse disponibili.
Nella vostra Regione ci sono state criticità sui pazienti non residenti o sulla gestione delle strutture private accreditate?
L’accesso dei pazienti fuori Regione in Emilia Romagna dipende dal tipo di patologia, dalla classe di farmaco e dalle competenze delle nostre strutture: per le patologie neoplastiche la mobilità proveniente da altre regioni è maggiore, mentre ad esempio per il nusinersen si tratta di pochissimi casi. In generale non abbiamo riscontrato alcuna criticità anche perché, su questo tema, l’organizzazione dei fondi per i farmaci innovativi prevede la separazione dei rimborsi tra residenti e non residenti. È un aspetto ben affrontato dalla normativa.
In Emilia Romagna il numero di strutture private accreditate è molto limitato e quindi meno problematico rispetto ad altre Regioni.
Ci sono criticità per il fatto che il fondo degli innovativi è finanziato a livello nazionale ed è gestito a livello regionale? Come si armonizzano questi due piani?
Questo è decisamente il punto più critico della gestione, soprattutto per quanto riguarda la rendicontazione. Nel caso dei farmaci per l’epatite C noi l’abbiamo affrontato prevedendo un piano terapeutico regionale informatizzato che i nostri clinici hanno dovuto compilare in aggiunta al piano terapeutico previsto da AIFA. Ciò si è reso necessario per disporre di informazioni dettagliate e tempestive sulla spesa regionale aggiornate in tempo reale, mese per mese per consentire di programmare la spesa e utilizzare al meglio tutti i fondi disponibili. I dati di monitoraggio AIFA infatti sono parziali e soprattutto i dati di spesa sono disponibili in maniera più completa solo a fine anno. Questa procedura ha richiesto un maggiore impegno da parte dei nostri clinici, e rappresenta un problema rispetto al quale bisognerà trovare una soluzione alternativa in futuro.
In questo modo noi abbiamo programmato la spesa ed effettuato i rendiconti ai clinici, alle Aziende Sanitarie e ai servizi farmaceutici, con cadenza mensile: ad esempio, ad ottobre 2017 siamo stati in grado di stimare, sulla base della prescrizione effettuata, una spesa di 28 milioni per l’anno 2017 a fronte di un fondo disponibile di 34 milioni, e questo dato ci ha consentito di rassicurare i clinici sulla capienza del fondo fino a fine anno invitandoli ad aumentare, compatibilmente con i problemi organizzativi, la prescrizione. Perché l’obiettivo rimane quello di spendere in maniera appropriata tutte le risorse a disposizione ma, in mancanza di dati certi sull’andamento della spesa, la gestione e la programmazione della prescrizione risulta difficile. A maggior ragione quando, come nel caso dei farmaci per l’epatite C, il sistema di definizione dei prezzi è veramente molto complicato e prevede anche regole di confidenzialità da rispettare: in questa situazione, a nostro parere, il ruolo della Regione è ancora più importante come garanzia di una informazione super partes, al di là delle aziende farmaceutiche.
Alla fine del 2017 il nostro dato di spesa finale per i farmaci innovativi non oncologici è stato di circa 32 milioni di euro. Dal monitoraggio AIFA relativo al periodo gennaio-novembre 2017, reso noto nella Riunione del Consiglio di Amministrazione AIFA del 29 marzo 2018 e che successivamente è stato aggiornato, la spesa per il 2017 per la Regione Emilia Romagna risultava essere di circa 7 milioni di euro. In realtà, nessuno dei due dati era sbagliato, si trattava di una diversa modalità di presentare i dati attribuendo temporalmente in modo diverso i rimborsi tramite payback. Quello che mi preme sottolineare in questo caso, fuor di ogni polemica, è che il report elaborato da AIFA, seppure corretto dal punto di vista contabile, non ci fornisce un aiuto concreto per il governo della spesa. Sarebbe opportuno quindi trovare una soluzione condivisa tra tutti gli attori coinvolti, in modo che le Regioni possano contare su un dato tempestivo, affidabile e utile per il controllo della spesa e per la programmazione delle prescrizioni da parte dei clinici. Considerando che per tutti i farmaci innovativi è stabilito l’obbligo della compilazione di un registro, sarebbe auspicabile predisporre un coordinamento tra il livello centrale e il livello regionale in modo tale che tutti possano accedere agli stessi dati in modo tempestivo senza la necessità di duplicare i registri e i database a livello regionale, evitando di appesantire il lavoro dei clinici e rendendo più efficienti le procedure.
Il commento di Roberta Di Turi
Direttore Dipartimento dei Servizi, Direttore UOC Assistenza farmaceutica, Asl Roma 3
Come viene gestito l’accesso al fondo dei farmaci innovativi nella sua Regione e la ripartizione tra le diverse aziende sanitarie?
Prima del 2017, in mancanza di una normativa dedicata, il tema non si poneva. Come noto, dal 1° gennaio 2017 sono stati istituiti i due fondi nazionali per il concorso al rimborso della spesa regionale effettuata per l’acquisto di medicinali innovativi, oncologici e non oncologici. Dal quel momento quindi, anche nella nostra Regione, eravamo a conoscenza che, in base alla normativa nazionale, avremmo avuto accesso e diritto a questi fondi ma fino al 28 dicembre 2017 la Regione Lazio non ha legiferato in tal senso. In quella data, sono stati emanati i provvedimenti che hanno disposto la ripartizione dei fondi, con una distinzione tra innovativi oncologici e non: per i farmaci non oncologici la ripartizione è stata prevista tra le sole aziende sanitarie locali sulla base dei consumi calcolati in proporzione ai residenti delle singole ASL. L’attribuzione dei fondi è stata però “virtuale”, nel senso che compare nel bilancio regionale ripartito per le diverse aziende ma non è stata seguita nei fatti da un travaso di risorse verso i cosiddetti “payers”, cioè gli operatori che hanno la responsabilità della spesa. In pratica, il conto delle farmacie interne alle ASL e dei Servizi farmaceutici territoriali è risultato invariato rispetto alla spesa effettuata: per questo motivo possiamo definirlo un ritorno “virtuale” al bilancio aziendale che però, di fatto, non è arrivato alle strutture periferiche competenti.
Il fondo per i farmaci innovativi oncologici è stato trattato in maniera diversa perché la delibera regionale ha previsto la ripartizione tra tutte le aziende sanitarie, comprese le aziende ospedaliere e gli Istituti di Ricovero e Cura a Carattere Scientifico (IRCCS). In questo caso, la Regione ha decretato che la ripartizione tenesse conto di chi effettivamente somministrava il farmaco, e quindi anche delle aziende ospedaliere. A mio parere, questa modalità di ripartizione è più corretta e sarebbe utile anche nel caso dei farmaci innovativi non oncologici: ad esempio, i medicinali anti HCV vengono dispensati da un numero ridotto di aziende ospedaliere ma, in una partita di giro, il ritorno economico è destinato alle singole ASL che invece, di fatto, non dispensano.
Comunque, anche nel caso degli oncologici, alcune aziende ospedaliere hanno registrato a bilancio la riassegnazione virtuale ma, ancora una volta, questa quota non è stata stornata dalla spesa attribuita alle Unità Operative che l’avevano prodotta, tantomeno è stata resa nota ai Servizi Farmaceutici.
E si tratta di cifre importanti: stiamo parlando di oltre 54 milioni di euro assegnati alla Regione Lazio per i farmaci oncologici innovativi e altrettanto per i farmaci innovativi non oncologici.
Quali criticità ha riscontrato maggiormente nella gestione pratica di questi fondi?
La non completa trasparenza delle procedure per l’accesso ai farmaci innovativi ha comportato una situazione di incertezza da parte dei payers ma anche dei clinici che, non sentendosi garantiti da una normativa regionale certa e tempestiva, hanno adottato un atteggiamento prescrittivo molto cauto nei confronti di questi farmaci: questo è accaduto nella nostra Regione ma dobbiamo ricordare come, anche a livello nazionale, i fondi stanziati dalla legge nazionale 2017 di stabilità non siano stati utilizzati del tutto.
Nonostante questa situazione di incertezza, ogni farmacista dirigente, nella sua realtà e struttura, si muove considerando le motivazioni etiche che sono alla base della professione, cercando quindi di garantire l’accesso ai farmaci a tutti i pazienti che ne hanno bisogno, nell’ambito della previsione di bilancio elaborata a inizio anno. Nella pratica, pur sapendo che esistono i fondi per i farmaci innovativi, spesso i payers regionali non sono messi in condizione di poterci fare affidamento. Il che, ovviamente, non rappresenta una modalità rassicurante né per gli operatori né tantomeno per i cittadini.
A livello di rendicontazione, in mancanza di procedure chiare da mettere in atto, i responsabili della spesa hanno agito in maniera autonoma e in base alla propria esperienza o necessità: per quanto riguarda la mia struttura, ad esempio, abbiamo registrato e messo in evidenza tutta la spesa complessiva legata agli innovativi, senza però averne nessun esito contabile in pratica.
La gestione del fondo, con erogazione a livello nazionale, e la prescrizione dei farmaci, che è a livello regionale, sono due strade che si armonizzano o questa doppia via può creare delle criticità?
Al di là degli obblighi normativi stabiliti a livello nazionale, come il monitoraggio da parte dell’AIFA, e l’obbligo di debito informativo regionale che le strutture devono assolvere, la gestione a livello regionale è lasciata all’iniziativa delle singole aziende che, anche in assetto multidisciplinare, agiscono però in base alla propria programmazione. Possiamo dire che, se la via nazionale è tracciata, la via regionale rimane in capo ai singoli: non c’è una direttiva regionale che rende i percorsi omogenei, con il rischio che anche l’accesso ai farmaci non sia omogeneo in quanto dipende dalle condizioni di bilancio delle singole aziende.
Questi fondi per i farmaci innovativi possono rappresentare un’opportunità o un limite?
Un limite sicuramente no, potrebbero rappresentare una grande opportunità. La prima esperienza fatta in tal senso ci ha dimostrato però che, in mancanza di indicazioni e orientamenti chiari da parte delle Regioni, questi fondi rischiano di essere degli strumenti poco utili. Se viene stanziato un finanziamento ad hoc, la logica vorrebbe che quel finanziamento andasse utilizzato fino in fondo, per coprire dei bisogni insoddisfatti e agevolare quelle fasce di popolazione più deboli che ne hanno bisogno. In questo momento, non sembra che questi risultati siano stati raggiunti appieno. La politica di scarsa trasparenza e assenza di provvedimenti certi a livello regionale rischia di inficiare gli investimenti e gli sforzi sostenuti a livello nazionale. Un vero peccato, secondo me.
Il commento di Daria Bettoni
Farmacista ospedaliera, ASST Spedali Civili di Brescia
Come viene gestito il fondo farmaci innovativi nella sua Regione e la ripartizione tra le diverse aziende sanitarie? Quali sono le direttive regionali?
Da sempre la politica sanitaria regionale della Lombardia è ispirata alla formalizzazione di linee di indirizzo e non di imposizione; i vincoli prescrittivi, previsti dalla normativa nazionale, vengono applicati tenendo conto anche dell’organizzazione ospedaliera e delle esigenze territoriali. Ciò ha costantemente consentito un accesso veloce ai nuovi farmaci. Ad esempio, non è presente un Prontuario Terapeutico Regionale, che di fatto, come più volte sottolineato da AIFA, rallenta significativamente la disponibilità delle terapie innovative nelle Regioni in cui è stato adottato.
In Lombardia sono frequentemente istituiti specifici gruppi di lavoro multidisciplinari, che coinvolgono i medici dei centri di riferimento per le diverse patologie e i farmacisti ospedalieri, affinché venga individuata la casistica (spesso tramite survey) e siano condivisi i percorsi di cura.
La Regione Lombardia, per ogni farmaco inserito nei registri di monitoraggio dell’AIFA (non solo per gli innovativi), individua gli specialisti prescrittori e le UO afferenti, privilegiando le scelta dell’approccio “multidisciplinare”. In questo modo si ottimizza il trattamento terapeutico, rispondendo alle necessità cliniche, gestionali e logistiche del paziente, tenendo conto anche delle differenti situazioni organizzative locali. Possono essere inoltre autorizzate “deroghe”, su richiesta dei singoli centri, che devono evidenziare la casistica clinica e particolari esigenze che possono migliorare la gestione dei pazienti.
La Regione Lombardia inoltre promuove lo sviluppo di reti del tipo hub e spoke, in cui gli specialisti condividono le scelte prescrittive, i dati di esito e la corretta distribuzione delle risorse e il monitoraggio della spesa farmaceutica. In una Regione come la Lombardia, con molti centri ospedalieri specialistici di eccellenza, di grande estensione territoriale e densamente popolata, questo modello è particolarmente efficace e garantisce una maggior appropriatezza. In particolare sono da tempo presenti reti oncologiche lombarde (REL e ROL) e per le malattie rare; sono state istituite reti per l’HCV: questa scelta organizzativa consente l’accesso alle terapie innovative in tempi brevi.
In merito alla specifica casistica dei farmaci innovativi, ad oggi la ripartizione del fondo rientra nel finanziamento complessivo dell’assistenza farmaceutica che viene formalizzato ad inizio di ogni anno dalla Regione a favore delle strutture sanitarie pubbliche e private accreditate. Non c’è quindi un fondo per Azienda, ma, come i biosimilari, sono scorporati dall’abbattimento del tetto di File F regionale con una sorta di rimborso “a piè di lista”.
Solo nel caso dei farmaci antiHCV sono assegnati specifici finanziamenti vincolanti, ma riprogrammabili in corso d’anno, e dedicati alle reti/singole strutture.
Ci sono procedure differenti per i farmaci oncologici e non oncologici?
Per talune tipologie di farmaci, per le malattie rare (es. atrofia muscolare spinale – SMA – e fibrosi cistica), sono istituiti percorsi che includono il riconoscimento dei centri di riferimento da parte del Centro di Coordinamento Regionale per le Malattie Rare, che promuove gruppi di lavoro multidisciplinari per l’elaborazione e l’aggiornamento dei PDTA; ad oggi sono 110 i PDTA per le malattie rare disponibili sul sito del Centro di Coordinamento Regionale: fra questi è presente quello per la SMA, in cui è stato incluso il trattamento con nusinersen.
Nell’ambito delle malattie rare le scelte delle singole Regioni sono condivise e coordinate in un Tavolo nazionale afferente alla Commissione Salute, a cui partecipo come referente per la Lombardia. In particolare per la nuova terapia per la SMA, che è particolarmente complessa per le modalità di somministrazione, il confronto e l’adozione di scelte concordate è fondamentale per rispondere alle esigenze dei pazienti che spesso si rivolgono anche a centri lontani dalla Regione di residenza.
In sintesi per farmaci innovativi per malattie rare sono identificati i centri autorizzati alla prescrizione.
La stessa situazione avviene per l’HCV, per cui, come già detto, sono individuati specifici centri autorizzati alla prescrizione. Inoltre, all’interno di tale elenco sono state istituite due reti inter-ospedaliere, fra loro connesse, la rete bergamasca e la rete bresciana, coordinate rispettivamente dall’ASST Papa Giovanni XXIII di Bergamo e l’ASST Spedali Civili di Brescia. Ogni rete fa riferimento ai suddetti, ma coinvolge più strutture e specialisti (infettivologi, gastroenterologi, epatologi e farmacisti), in modo da avere una costante evidenza delle esigenze cliniche, delle terapie in corso e già concluse, degli esiti, oltre che della spesa farmaceutica sostenuta e delle risorse necessarie per l’attuazione delle terapie programmate.
Il budget assegnato è determinato sulla base di survey periodicamente aggiornate, tenendo anche conto della variabilità dei costi dei farmaci, dei nuovi casi, delle terapie concluse e dei rimborsi pervenuti, e può comunque essere ridiscusso e rivalutato, se necessario.
Differente è la situazione dei farmaci oncologici per i quali, a causa della numerosità della casistica, ad oggi si è preferito autorizzare tutte le strutture di oncologia pubbliche e private accreditate (salvo specifici farmaci o indicazioni terapeutiche in cui vengono individuati specifici centri).
Per i farmaci oncologici la Regione ha un rapporto diretto, oltre che con i farmacisti ospedalieri, con le reti di riferimento oncologiche (ROL e REL), che sono da tempo diffuse, come in altre Regioni, ad esempio Piemonte, Toscana, Trentino, Umbria, Veneto.
Come viene gestito il privato accreditato?
Il privato accreditato rispetta le stesse regole del pubblico, i centri devono essere specificatamente autorizzati, in coerenza con la logica della libertà di scelta del paziente, che deve poter essere curato dagli specialisti a cui si è affidato.
Il DM del 16 febbraio 2018 prevede che l’acquisto sia effettuato da centri ospedalieri pubblici, per questo i farmaci per l’HCV sono acquistati dalle ASST pubbliche di riferimento, specificatamente individuate dalla Regione, che, con una gestione “in rete”, provvedono all’acquisto e all’erogazione agli ospedali privati autorizzati, al monitoraggio previsto dai registri AIFA e alla gestione del budget complessivo.
Si tratta di un sistema complesso, reso necessario dalla disposizione ministeriale, che può rendere poi difficile la corretta tracciabilità del dato a livello dei registri di monitoraggio, per questo è stata recentemente inserita da AIFA la nuova funzionalità di identificazione della natura pubblica o privata della struttura acquirente. Purtroppo uno sviluppo selettivo che consenta la classificazione solo per i registri innovativi non è possibile a causa della configurazione integrata della piattaforma.
Per i farmaci innovativi non oncologici gli acquisti già vengono effettuati da strutture pubbliche, anche se erogati da privati accreditati, mentre nel caso degli oncologici innovativi Regione Lombardia sta organizzando un sistema di acquisto analogo al fine di non perdere l’accesso al fondo innovativi, in quanto la spesa sostenuta dai privati accreditati al momento è a carico del bilancio regionale.
Nella sua struttura avete anche casi di pazienti da fuori Regione? Come vengono gestiti?
Il nostro ospedale, come molti della Regione Lombardia, è un centro “attrattivo” anche per pazienti non bresciani, spesso provenienti da altre Regioni. L’indicazione della Regione Lombardia è sempre stata di massima disponibilità, per cui le terapie vengono prescritte ed erogate, nel rispetto della libertà di scelta del paziente.
Analogamente, i pazienti lombardi possono essere seguiti, anche per i farmaci innovativi, presso ospedali non lombardi. La gestione, concordata con i centri di riferimento, può essere interamente a carico del centro prescrittore oppure il farmaco può essere dispensato anche dalla farmacia di residenza, come è consentito dal sistema dei registri AIFA.
In taluni casi (HCV), solo per alcune Regioni, la compensazione non è automatica, ma va specificatamente richiesta, tramite la ASL di residenza, alla Regione di residenza.
La spesa relativa ai non residenti è rimborsata alle Regioni mediante la fatturazione diretta fra enti del SSN.
Esistono criticità sulla rendicontazione o il monitoraggio?
Le attività di rendicontazione e monitoraggio, fra loro strettamente correlate, si estendono a tutti i farmaci inclusi nei registri AIFA, ad oggi sono 132 i registri e i piani terapeutici web based. Ciò comporta molto lavoro per i centri ospedalieri da parte dei medici prescrittori e dei farmacisti ospedalieri, sia per la compilazione dei registri AIFA, che per la valutazione del bisogno clinico mediante survey, la rendicontazione mediante File F, l’elaborazione dei periodici report sulla spesa farmaceutica, indispensabili per la valutazione dei budget da assegnare a livello regionale. Inoltre sono previste periodiche e capillari verifiche da parte delle Agenzie di Tutela della Salute (ATS) su queste attività.
L’informazione di ritorno da parte di AIFA potrebbe e dovrebbe essere più tempestiva: sono pubblicati dei report mensili e annuali sulla spesa farmaceutica nazionale e regionale, ma dopo diversi mesi (ad es. il Monitoraggio della Spesa Farmaceutica Nazionale e Regionale di gennaio-aprile 2018 è del 24 settembre 2018), mentre il rapporto Osmed 2017 è stato reso disponibile a luglio 2018.
Il carattere nazionale del finanziamento del fondo si armonizza con l’erogazione dei farmaci, che avviene a livello regionale?
Attualmente, dal 2018, la gestione dei fondi è mista e prevede un acconto per le Regioni sulla base del fabbisogno indistinto, stabilito dall’accordo Stato-Regioni, considerando anche il conguaglio 2017, con un saldo relativo alla spesa effettivamente sostenuta per i pazienti residenti. La mancanza di una chiara ripartizione regionale dei fondi per i farmaci innovativi oncologici e non oncologici, che potrebbe essere effettuata sulla base della spesa farmaceutica complessiva regionale, quando non esistano differenze territoriali di incidenza e prevalenza per le patologie trattate, rende più complessa la gestione delle risorse a livello regionale ed espone le Regioni al rischio di un mancato rimborso.
Inoltre, oltre alle informazioni sulla spesa farmaceutica, andrebbero pubblicati dati epidemiologici, così come i risultati dei trattamenti conclusi. L’ambito in cui è presente una reportistica puntuale è quello dell’HCV, questo modus operandi andrebbe esteso anche a tutti gli altri farmaci innovativi.
Ma la vera criticità è costituita dal ritardo con cui AIFA pubblica i dati di accesso ai fondi da parte delle Regioni, cosa che impedisce a queste ultime una reale azione di programmazione.
Un commento generale sul fondo farmaci innovativi: quali sono le opportunità o i limiti? Si potrebbe migliorare qualche aspetto?
Come evidenziato nel Position Statement di GIMBE, sarebbe necessaria una maggior trasparenza nei criteri di valutazione adottati per decidere l’innovatività di un farmaco. Le basi scientifiche e metodologiche sono condivisibili, ma il metodo di valutazione non è stato esplicitato per quanto riguarda le variabili considerate: bisogno terapeutico, valore terapeutico aggiunto e qualità delle prove.
Inoltre il fatto che i fondi effettivamente spesi per i farmaci innovativi e non innovativi siano molto inferiori alle risorse stanziate significa che il sistema di gestione attuale non è ottimale e potrebbe essere migliorato l’accesso dei pazienti a terapie di riconosciuta efficacia.
Gli obiettivi strategici del nostro Servizio Sanitario Nazionale sono la qualità delle cure, l’equità e la sostenibilità. Per il loro conseguimento è essenziale non circoscrivere le valutazioni ai trattamenti farmacologici, ma definire e condividere, a livello nazionale, valutazioni di Health Technology Assessment e percorsi diagnostici, terapeutici e assistenziali (PDTA), creare gruppi di lavoro multidisciplinari di riferimento, selezionare i centri prescrittori individuando le eccellenze, promuovere, sul modello delle Malattie Rare, la diffusione delle reti di patologia, migliorare l’integrazione dei meccanismi di programmazione e controllo. I finanziamenti dovrebbero poter essere adeguati alla valutazione degli esiti delle terapie, ciò è possibile adottando strumenti informativi che correlino l’appropriatezza prescrittiva e l’efficienza economica all’efficacia dei trattamenti, mettendo in relazione i dati clinici disponibili dai registri AIFA con quelli amministrativi sui farmaci (File F), e delle prestazioni (schede di dimissione ospedaliera – SDO).
La politica del farmaco in Italia è, da diverso tempo, influenzata dalla presenza di tetti sulla spesa farmaceutica [Jommi & Minghetti, 2015].
Introdotti dalla Legge 405/2001 e riferiti inizialmente alla sola spesa farmaceutica territoriale, tali tetti sono stati modificati per la prima volta dalla Legge 326/2003, che ha definito, come “valore di riferimento” per la spesa complessiva per farmaci, il 16% della spesa sanitaria pubblica. È però la Legge 222/2007 ad avere introdotto:
un doppio tetto; uno, pari al 14% delle risorse assegnate al Servizio Sanitario Nazionale (SSN), riferito alla spesa per farmaci utilizzati sul territorio, includendo i farmaci acquistati e dispensati dalle farmacie aperte al pubblico (spesa convenzionata) e i farmaci in Fascia A distribuiti direttamente dalle aziende sanitarie o dalle farmacie aperte al pubblico per conto del SSN; l’altro, pari al 2,4% delle risorse SSN, per i farmaci utilizzati in ospedale;
il ripiano dell’eventuale sfondamento del tetto sulla territoriale da parte dei tre attori della filiera (industria, grossisti e farmacie);
la definizione di un budget per le imprese farmaceutiche, basato essenzialmente sul fatturato dell’ultimo anno, con successiva allocazione dell’eventuale payback sulla base del contribuito di ciascuna impresa allo sfondamento del tetto.
La Legge 135/2012 ha previsto, a partire dal 2013, una riduzione del tetto sulla territoriale all’11,35% e un aumento di quello sull’ospedaliera al 3,5%; la stessa legge ha introdotto una compartecipazione dell’industria del 50% all’eventuale sfondamento del tetto sull’ospedaliera, sempre in proporzione al contributo di ciascuna azienda allo sfondamento del tetto. Infine, la Legge 232/2016 ha modificato dal 2017 la struttura dei tetti prevedendone uno per la spesa convenzionata (7,96%, delle risorse SSN) e uno per tutti i farmaci acquistati dalle aziende sanitarie (6,89%).
Tetti e fondi presentano diversi elementi di criticità
Ai tetti “ordinari” si aggiungono i fondi per l’innovazione. La Legge 190/2014 ha istituito un fondo ad hoc per i medicinali innovativi, pari a 500 milioni a valere sul 2015 e il 2016. Tale fondo è stato prevalentemente utilizzato per l’acquisto di farmaci per l’epatite C. La Legge 232/2016 ha previsto l’istituzione di due fondi, con una dotazione di 500 milioni ciascuno, uno per medicinali innovativi e uno per i farmaci oncologici innovativi, attingendo in parte a risorse già assegnate al SSN per obiettivi del Piano Sanitario Nazionale. I farmaci innovativi, oltre alla disponibilità di fondi dedicati, hanno accesso immediato ai mercati regionali (in linea di principio, il loro utilizzo non è condizionato all’inclusione nei prontuari regionali), non partecipano (con i farmaci orfani) alla copertura di eventuali sfondamenti dei tetti di spesa e non sono soggetti ad uno sconto obbligatorio del 5% + 5% del prezzo negoziato con AIFA (Agenzia Italiana del Farmaco), previsto per tutti i farmaci coperti dal SSN.
I criteri per la valutazione dell’innovatività, in capo alla CTS (Commissione Tecnico-Scientifica) dell’AIFA, sono stati definiti dalla Determina 519/2017 e si tratta del livello di bisogno terapeutico, del valore terapeutico aggiunto e della qualità delle evidenze prodotte negli studi registrativi. L’elenco dei farmaci innovativi dovrebbe essere aggiornato mensilmente sul sito dell’AIFA (al 31 luglio, l’ultimo aggiornamento è del 16/7/2018). La principale novità, rispetto alla precedente gestione dell’innovatività basata sul documento CTS del 10/7/2010 (“Criteri per l’attribuzione del grado di innovazione terapeutica dei nuovi farmaci”) è la pubblicazione sul sito AIFA di schede di “appraisal” con cui viene descritto il razionale della valutazione da parte della CTS. Delle 18 richieste di innovatività per le quali è stato pubblicato l’esito, 6 sono state accolte (per un farmaco si tratta di innovatività “di classe”, ovvero innovatività collegata a quella acquisita da precedenti farmaci caratterizzati dallo stesso meccanismo di azione), a 4 farmaci è stata attribuita l’innovatività condizionata alla produzione entro 18 mesi di ulteriori evidenze a supporto della richiesta (i farmaci ad innovatività condizionata hanno solo il vantaggio dell’accesso immediato ai mercati regionali) e 8 sono stati giudicati non innovativi.
Figura 1. Spesa farmaceutica SSN e spesa totale SSN (netto farmaci) (2001-2017; 2001=100)
Fonte: elaborazione Osservatorio Farmaci Cergas Bocconi su dati Conti Economici di Aziende Sanitarie e AIFA
CAGR = Compound Annual Growth Rate
Il sistema dei tetti ha permesso di definire un quadro complessivo di risorse da destinare ex ante alla farmaceutica. La presenza ex post di sistemi di compartecipazione allo sfondamento dei tetti ha generato aspettative di “sostenibilità” dell’incremento della spesa. Le regioni, al di là di una responsabilizzazione complessiva sulla spesa sanitaria, vengono poi valutate anche sul posizionamento rispetto ai tetti di spesa farmaceutica, e sono state quindi spinte ad adottare politiche di razionalizzazione e, in diversi casi, di contenimento, della spesa farmaceutica. Non è un caso che la spesa complessiva per farmaci a carico del SSN (al lordo peraltro dei diversi payback da parte delle imprese) sia aumentata in media del 2% dal 2001 al 2017 a fronte di un, pur contenuto, 2,7% di crescita media della spesa sanitaria pubblica (Figura 1).
È possibile superare la logica di gestione a silos della spesa farmaceutica?
I fondi per farmaci innovativi hanno avuto un ruolo importante, segnaletico della volontà di garantire risorse per farmaci prioritari per il SSN, aumentando il livello di trasparenza sulle scelte di identificazione dei farmaci innovativi.
Tuttavia, tetti e fondi presentano diversi elementi di criticità.
I tetti si ispirano ad una logica di gestione a silos della spesa e non considerano gli effetti dei farmaci su altre prestazioni a carico del SSN (ad esempio, minori costi per ricoveri evitati o maggiori costi per la gestione degli effetti collaterali). Vi è quindi un’incoerenza di fondo tra un interesse crescente per la gestione integrata del paziente e per i percorsi diagnostico-terapeutici assistenziali da una parte, e la definizione di tetti di spesa per fattori produttivi dall’altra. È vero che la Legge 205/2017 ha previsto un monitoraggio in via sperimentale, per il triennio 2018-2020, degli effetti dell’utilizzo dei farmaci innovativi sul costo del percorso terapeutico-assistenziale, richiamando quindi esplicitamente l’importanza di valutare gli effetti (positivi o negativi) di un farmaco sul costo del percorso complessivo del paziente. Ed è anche vero che l’obiettivo dichiarato di tale monitoraggio è di «migliorare l’allocazione delle risorse programmate per il SSN» e «valutare la congruità dei fondi per i farmaci innovativi». Rimane però l’approccio silos, in quanto non sono ancora chiari gli effetti attesi di tale analisi.
In secondo luogo, fissare un tetto come quota delle risorse a carico del SSN significa non tenere conto dell’evoluzione tendenziale della spesa farmaceutica, che dipende da dinamiche specifiche: dall’introduzione di nuovi farmaci, dalla scadenza dei brevetti e dalla variazione del mix prescrittivo nel tempo. Non esiste alcun Paese, tra i principali europei, ad avere adottato un tetto sulla farmaceutica commisurato alle risorse per la sanità pubblica.
Nel triennio 2013-2015 il payback effettivo è stato pari all’85% di quello potenziale
Il tetto sulla spesa ospedaliera e, dal 2017, quello sui farmaci acquistati dalle aziende sanitarie, è stato fissato ad un valore inferiore alla spesa consuntivata l’anno precedente, mentre quello più recente sulla convenzionata è superiore alla spesa consuntivata nell’anno precedente. Poiché è noto che la spesa per acquisti cresca più della convenzionata (che, anzi, è calata sistematicamente negli ultimi dieci anni), questo significa avere fissato dei tetti che predeterminano già degli squilibri (uno un disavanzo, l’altro un surplus).
Non viene poi prevista alcuna compensazione tra disavanzo sul tetto per farmaci acquistati dalle aziende sanitarie (1.580 milioni di euro circa nel 2017, secondo l’ultimo monitoraggio AIFA disponibile – consultato il 27 luglio 2018) e surplus su tetto della farmaceutica convenzionata, surplus presente anche per i fondi per farmaci innovativi (complessivamente pari a 920 milioni di euro, tra tetto sulla convenzionata – 480 milioni di euro, fondo per gli innovativi oncologici – 90 milioni di euro, e fondo per gli innovativi non oncologici – 360 milioni di euro). Visto l’andamento tendenziale della spesa per acquisti e di quello per la spesa convenzionata, il fenomeno si amplificherà nei prossimi anni.
Il livello di complessità nella gestione dei tetti e la scarsa trasparenza (in particolare sul flusso di spesa per farmaci a distribuzione diretta e per conto) hanno prodotto un aspro contenzioso tra industria e SSN, con il risultato che i tempi di pagamento da parte delle imprese si sono protratti nel tempo e che sono state offerte scontistiche dall’SSN sul payback dovuto: nel triennio 2013-2015 il payback effettivo è stato pari all’85% di quello potenziale. E nessuno ha ad oggi stimato il costo della gestione del contenzioso tra industria e SSN.
Inoltre, la distribuzione del payback tra le singole imprese, per quanto ispirata a logiche di equità (compartecipazioni in base al contributo alla crescita del mercato) non è avulsa da elementi di criticità. In primo luogo, lo sfondamento attribuibile ai farmaci innovativi e agli orfani viene riversato sugli altri prodotti coperti da brevetto. Essendo poi il budget per azienda costruito a partire dal mercato degli ultimi 12 mesi, i nuovi prodotti lanciati sul mercato (a meno che non siano innovativi o orfani) sono stati storicamente penalizzati, in quanto contribuiscono in modo rilevante alla crescita, non avendo alcun fatturato pregresso. Per questo la Legge 205/2017 ha previsto che le imprese paghino nel primo anno solo un contributo del 10% del fatturato realizzato per i nuovi farmaci, riallocando però il restante 90% su tutti gli altri farmaci, esclusi orfani e innovativi (ma inclusi generici e biosimilari).
Infine, l’assegnazione dei fondi per i farmaci innovativi è avvenuta con grande ritardo: solo nel 2018 (con Decreto del Ministero della Salute del 16 febbraio 2018, pubblicato in Gazzetta Ufficiale il 7/4/2018) sono state definite le modalità operative di erogazione alle regioni delle risorse stanziate per i farmaci innovativi (oncologici e non) per il 2017 (e il 2018).
È chiaro che in prospettiva il sistema dei tetti così definito non può reggere.
Tetto unico per la farmaceutica e target di crescita della spesa negoziato sono tra le proposte dell’Osservatorio Farmaci del Cergas Bocconi
Una proposta organica di revisione dei tetti è stata pubblicata a fine 2016 dall’Osservatorio Farmaci del Cergas Bocconi [Amoroso et al, 2016]. In particolare, si è suggerita una semplificazione dei tetti in una prima fase articolandoli in modo più coerente con la robustezza del flussi informativi disponibili; tale proposta è stata implementata dalla Legge 232/2016, che ha accorpato in un unico tetto tutti gli acquisti di farmaci da parte delle aziende sanitarie, indipendentemente dalla loro destinazione (utilizzo in ambito ospedaliero o in distribuzione diretta / per conto); in una seconda fase, unificando i due tetti sulla spesa convenzionata e sulla spesa per acquisti di farmaci dalle aziende sanitarie, per superare i problemi generati dalla mancata compensazione tra eventuali surplus e disavanzi. Guardando ad un orizzonte temporale più lungo, si è però suggerito di:
abbandonare il tetto di spesa e trasformarlo in target di crescita della spesa a tre/cinque anni, target rinegoziato annualmente tra le principali rappresentanze dell’industria e il Ministero della Salute e dell’Economia;
affidare ad AIFA un ruolo di supporto tecnico a tale negoziazione, che si sostanzierebbe in una stima dell’effetto atteso dell’ingresso sul mercato di nuovi prodotti, della scadenza dei brevetti, della modifica del mix di prescrizione di terapie già disponibili sul mercato, e degli eventuali effetti attesi dei farmaco su altre prestazioni sanitarie. Ciò peraltro spingerebbe AIFA a rafforzare ulteriormente la propria attività di horizon scanning, ai fini di una corretta programmazione della spesa.
Una seconda riflessione riguarda i fondi per i farmaci innovativi. Con tali fondi, come già osservato, la politica ha chiaramente segnalato la propria volontà di trovare risorse per farmaci considerati prioritari per il SSN. La conseguenza dei fondi è stata però quella di creare silos “protetti” all’interno di una logica silos. Sarebbe auspicabile invece che i fondi venissero incorporati nei tetti, mantenendo gli altri aspetti che facilitano l’accesso dei pazienti ai farmaci innovativi.
Se le proposte venissero attuate (tetto unico, compresi i fondi per gli innovativi, nel breve periodo; target di crescita nel medio-lungo periodo), sarebbe comunque necessario trovare una soluzione per il payback pregresso e non ancora pagato. A questo proposito è utile osservare che se si fosse applicato nel 2017 un meccanismo compensativo tra tetti di spesa e fondi, lo sfondamento del tetto per acquisti di farmaci da parte di aziende sanitarie si sarebbe ridotto da 1.580 milioni di euro a 660 milioni di euro, di cui 330 a carico dell’industria; se il trend della spesa complessiva per farmaci a carico del SSN fosse in futuro simile a quello registrato in media dal 2001, complessivamente lo sfondamento del tetto complessivo non dovrebbe aumentare in modo sensibile.
Con i fondi per gli innovativi la politica ha inteso rendere disponibili risorse aggiuntive per farmaci prioritari, ma sempre in una logica a silos
Sarebbe utile, alla luce di queste riflessioni, definire un accordo transattivo sui payback pregressi che ne riduca l’impatto complessivo; prevedere delle iniziative di contenimento, per abbattere la spesa a valori simili a quelli del tetto complessivo, iniziative già proposte in un articolo pubblicato nel 2015 [Martini, 2015]; avviare da subito un passaggio ad un tetto unico e, in prospettiva, ad un target di crescita della spesa negoziato, come già l’Osservatorio Farmaci aveva proposto.
Bibliografia di riferimento
Amoroso N, Armeni P, Costa F, et al. Il governo dell’assistenza farmaceutica in Italia: possibili traiettorie di cambiamento in Rapporto OASI 2016. Cergas – SDA Bocconi, 2016
Aru C, da Empoli S, Della Porta MR, et al. Inside-out: l’impatto dell’innovazione farmaceutica su spesa sanitaria e costi sociali e previdenziali. Istituto per la Competitività (I-Com), 2018
Jommi C, Minghetti P. Pharmaceutical Pricing Policies in Italy. In: Zaheer-Ud-Din Babar (ed.). Pharmaceutical Prices in the 21st Century. London: Springer, 2015, pp. 131-51
Martini N. Il settore farmaceutico rischia il default. Proposte per ristabilire l’equilibrio economico e per definire una nuova governance strutturale. Global & Regional Health Technology Assessment 2015; 2: 53-8
Intervista a Patrizia Popoli
Direttore del Centro Nazionale Ricerca e Valutazione preclinica e clinica dei Farmaci (CNRVF) dell’Istituto Superiore di Sanità e Presidente della Commissione Tecnico Scientifica (CTS) dell’AIFA
Qual è il suo bilancio di questo primo anno di applicazione della normativa che individua i criteri per la definizione dell’innovatività?
Dai commenti che abbiamo ricevuto e dai confronti con i diversi stakeholders, anche nel corso di convegni nei quali si è trattato questo tema, possiamo dire che complessivamente la novità è stata accolta con favore: abbiamo registrato una reazione positiva soprattutto rispetto all’approccio adottato, improntato ad una maggiore trasparenza rispetto al passato, grazie alla definizione e pubblicazione dei criteri per l’attribuzione del carattere di innovatività. In generale, il metodo applicato è stato apprezzato ed è risultato piuttosto condiviso da tutti, anche se, ovviamente, esistono sempre margini di miglioramento. Anche di questi aspetti si è discusso molto, e penso che alcune riflessioni potrebbero essere utili per la prossima Commissione.
Quali sono gli elementi che hanno suscitato qualche criticità o qualche riflessione, anche per il futuro?
Innanzitutto, un elemento che molto spesso le aziende farmaceutiche hanno fatto notare riguarda la terminologia utilizzata nelle valutazioni: per i criteri di bisogno terapeutico e valore terapeutico aggiunto sono stabiliti dei livelli basati su giudizi espressi in scala, da “massimo” a “scarso” o “assente”. Alcune definizioni di questi livelli possono essere interpretate in maniera errata. Per esempio, se per un farmaco il giudizio relativo al valore terapeutico aggiunto risulta essere “scarso” non si deve intendere che il valore terapeutico del farmaco sia “scarso” in senso assoluto, ma semplicemente che, quando lo compariamo all’alternativa terapeutica corrispondente di cui già disponiamo, questo farmaco offre sì dei vantaggi ma solo di scarsa entità. Quindi, non solo il farmaco è ammesso alla rimborsabilità, ma gli viene riconosciuto comunque un vantaggio rispetto all’esistente. È comprensibile che, quando si esprime un valore di “scarso”, questo possa essere interpretato non correttamente estendendolo al valore terapeutico intrinseco del nuovo farmaco, ma così non dovrebbe essere. E per questa ragione abbiamo sempre cercato, in ogni occasione in cui se ne è parlato, di chiarire questo potenziale equivoco. Una possibilità per evitare tali fraintendimenti potrebbe essere sostituire i giudizi con un’indicazione numerica corrispondente, come ad esempio una scala con valori di livello da 1 a 5.
Alcuni hanno invece avanzato dei commenti in merito all’innovatività per i farmaci oncologici, facendo notare che sarebbe stato utile stabilire dei criteri maggiormente dettagliati non solo per i diversi tipi di tumore ma anche per le diverse linee terapeutiche. Al momento non abbiamo previsto dei criteri diversi per i farmaci oncologici perché ritenevamo che questo non fosse corretto, in quanto ci sembrava giusto adottare un principio comune per tutti i farmaci; abbiamo però inserito delle note esplicative in merito agli indicatori di efficacia in oncologia, per evidenziare gli aspetti maggiormente importanti per questa tipologia di farmaci. La necessità di un maggiore livello di dettaglio sulle linee terapeutiche in oncologia potrebbe essere in effetti valutata in futuro.
La qualità delle prove attualmente viene giudicata tramite il metodo GRADE, che prevede delle indicazioni abbastanza rigide: un altro suggerimento che abbiamo ricevuto è stato quello di interpretare questo giudizio sul valore delle prove in maniera un po’ più elastica.
Una riflessione scaturita invece all’interno della Commissione, e che ha portato ad alcune modifiche del nostro modo di procedere già nel corso di questi ultimi mesi, ha riguardato la necessità di giustificare con maggiore dettaglio i giudizi, elaborando una relazione più estesa che spiegasse le ragioni alla base di una determinata decisione. Ad esempio, per rendere conto pienamente del bisogno terapeutico esistente in una specifica patologia è necessario illustrare in maniera esaustiva quali sono le opzioni terapeutiche a disposizione e soprattutto qual è il livello di efficacia dei farmaci disponibili. Se pensiamo al morbo di Parkinson, ad esempio, esiste già un certo numero di farmaci disponibili, che tuttavia sono efficaci solo sulla riduzione dei sintomi, e non “curano” la malattia: in questo caso, come in altri, il bisogno terapeutico rimane alto pur in presenza di farmaci in commercio. Analogamente, per il valore terapeutico aggiunto vanno riportate tutte le evidenze che sono state considerate per misurare il valore terapeutico della molecola in analisi e per il confronto con le alternative. È opportuno esplicitare meglio quali sono gli studi, quali le evidenze, quali i parametri presi in considerazione. Come dicevo, la Commissione ha già adottato un maggiore livello di dettaglio nelle sue relazioni ma sicuramente è un aspetto da tenere ben presente e cercare di migliorare ancora.
Un suggerimento avanzato da più parti, anche pubblicamente nel corso di eventi, ma al quale abbiamo deciso di non dare seguito è stato il collegamento tra la valutazione dell’innovatività e la definizione del prezzo del farmaco. Al momento abbiamo deciso di voler tenere disgiunti i due aspetti ritenendo che la valutazione dell’innovatività fosse di pertinenza tecnico-scientifica e che quindi dovesse prescindere dal costo del farmaco.
Per quanto riguarda l’eventualità di assegnare un “premium price” al farmaco riconosciuto come innovativo, in analogia con quanto avviene in altri Paesi, di fatto nel nostro sistema il farmaco innovativo viene già “premiato” con vantaggi economici, e anche consistenti: innanzitutto perché non è sottoposto all’obbligo di sconto del 5%+5% ma, aspetto ancora più rilevante, perché l’azienda farmaceutica non partecipa al ripiano della spesa in caso di sfondamento.
Nella sua esperienza, quale dei 3 criteri può essere maggiormente correlato all’attribuzione del carattere di innovatività?
Ovviamente tutti e tre i criteri devono essere soddisfatti: devono essere presenti il bisogno terapeutico e il valore terapeutico aggiunto, e le prove che sono state portate dall’azienda a dimostrazione della propria tesi devono essere robuste. Però è il valore terapeutico aggiunto l’elemento maggiormente importante, fermo restando che anche gli altri due criteri devono essere sempre a livelli alti. Ovviamente, con una qualità delle prove bassa, a meno che non si tratti di una patologia rara, non si può riconoscere l’innovatività perché viene meno tutta l’impostazione di base: se le prove non sono affidabili, non possiamo parlare di innovatività.
Per concludere, le chiediamo un commento sull’utilizzo dei fondi nei 2017 e per i prossimi anni.
I fondi assegnati per il 2017 non sono stati usati per intero e questo è dipeso, in grandissima parte, dal fatto che si è iniziato a lavorare sull’innovatività tardi, nel corso dell’anno. I criteri sono stati pubblicati a inizio aprile 2017 e da lì sono cominciate le domande delle aziende. Anche nei casi in cui la Commissione si è espressa in tempi molto rapidi, come ad esempio per Spinraza, i tempi per il completamento della procedura sono comunque piuttosto lunghi: una volta che la Commissione concede l’innovatività, il farmaco passa al Comitato Prezzi e Rimborso per la negoziazione con l’azienda e, raggiunto l’accordo, viene effettuata la pubblicazione sulla Gazzetta Ufficiale. Dal quel momento inizia l’iter per l’inserimento nei Prontuari Terapeutici Regionali. È evidente che sono necessari alcuni mesi per completare tutto il processo per cui, nel 2017, l’applicazione dei criteri e della normativa è potuta avvenire solo nella seconda metà dell’anno. A mio parere, difficilmente si sarebbero potuti spendere tutti i fondi assegnati al 2017: sicuramente, se fosse stata prevista la disponibilità per 12 mesi di anno solare, la situazione sarebbe stata diversa. Dalle proiezioni già disponibili per i primi mesi del 2018, sembra che i fondi dovrebbero essere occupati per intero quest’anno, una volta andati a regime. Rimangono comunque i tempi lunghi per il completamento di tutto l’iter.
A marzo 2016 l’Agenzia Europea del Farmaco (EMA) ha lanciato PRIME (PRIority Medicines scheme), un programma nato con l’obiettivo di sostenere lo sviluppo di farmaci in patologie dove i pazienti presentano dei bisogni terapeutici insoddisfatti. Tramite PRIME, l’EMA offre supporto alle aziende per la generazione di dati affidabili su efficacia e sicurezza di un farmaco al fine di velocizzare il processo di valutazione e l’accesso al mercato.
Nella stessa direzione si è mossa l’Italia, con l’emanazione di leggi e decreti per accelerare l’accesso ai farmaci con evidente valore terapeutico e che permettono di far fronte a bisogni insoddisfatti.
L’Articolo 1 (commi 400 e 401) della Legge di Bilancio 2017 (n. 232/2016) ha istituito due fondi, di 500 milioni di Euro ciascuno, per il concorso al rimborso alle Regioni per l’acquisto dei medicinali innovativi e dei medicinali oncologici innovativi.
Nel Box 1 sono riportati i commi della legge che stabiliscono le modalità di finanziamento dei Fondi, ai quali hanno accesso solo i farmaci ai quali viene riconosciuta innovatività piena.
La Legge di Bilancio 2017 ha istituito due fondi per il concorso al rimborso alle Regioni per l’acquisto dei medicinali innovativi
I farmaci a innovatività condizionata, invece, non hanno accesso ai fondi, ma vengono inseriti immediatamente nei Prontuari Terapeutici Regionali, come previsto dal Decreto-legge n. 158 del 13 settembre 2012 (convertito con modificazioni, dalla Legge n. 189 dell’8 novembre 2012). La Legge n. 232/2016 differenzia inoltre i farmaci innovativi e a innovatività condizionata per quanto riguarda la durata del requisito di innovatività che è, rispettivamente, di 36 e 18 mesi, mentre per tutti i farmaci interessati è previsto il monitoraggio dei registri AIFA.
Con quanto riportato nel comma 405, la Legge n. 232/2016 lascia al Ministero della Salute di concerto con il Ministro dell’Economia e delle Finanze la definizione delle modalità di erogazione dei fondi. Il 21 dicembre 2017 la Conferenza Permanente per i rapporti tra lo Stato, le Regioni e le province autonome di Trento e Bolzano ha approvato il Decreto che sancisce l’intesa sullo schema di decreto disciplinante le modalità operative di erogazione delle risorse di cui ai commi 400 e 401 della Legge n. 232/2016. In particolare, viene definito che l’accesso ai fondi per i farmaci innovativi è previsto per tutte le Regioni a eccezione di Friuli Venezia Giulia, Valle d’Aosta, Sardegna, province autonome di Trento e Bolzano e della Regione Sicilia che aderisce solo al 50%. Inoltre, vengono definiti criteri e modalità differenti a seconda che i pazienti trattati siano o meno residenti nella Regione in oggetto.
Per identificare le modalità di riconoscimento dell’innovatività, e il conseguente accesso ai fondi, nell’art.1 comma 402 veniva richiesto all’Agenzia Italiana del Farmaco (AIFA) di stabilire i criteri per la classificazione dei farmaci innovativi (oncologici e non).
Con la Determina n. 519/2017 del 31 marzo 2017, successivamente aggiornata e sostituita dalla Determina 1535/2017 del 12 settembre 2017, l’AIFA ha recepito quanto previsto dalla Legge di Bilancio 2017 e ha stabilito i criteri per la classificazione e la procedura di valutazione per la permanenza del requisito dell’innovatività. Gli obiettivi principali della normativa sono garantire e armonizzare a livello nazionale il rapido accesso ai farmaci che dimostrano un chiaro valore terapeutico aggiunto rispetto alle alternative disponibili e incentivare lo sviluppo di farmaci che offrano sostanziali benefici terapeutici per i pazienti.
Box 1. Legge 11 dicembre 2016 n. 232. Art. 1. Disposizioni in merito ai farmaci innovativi
Comma 400. A decorrere dal 1° gennaio 2017, nello stato di previsione del Ministero della Salute è istituito un Fondo per il concorso al rimborso alle regioni per l’acquisto dei medicinali innovativi, con una dotazione di 500 milioni di euro annui. Tale Fondo è finanziato rispettivamente per 325 milioni di euro per l’anno 2017, 223 milioni di euro per l’anno 2018, 164 milioni di euro a decorrere dall’anno 2019, mediante utilizzo delle risorse del comma 393 del presente articolo, e per 175 milioni di euro per l’anno 2017, 277 milioni per l’anno 2018, 336 milioni di euro a decorrere dall’anno 2019, mediante utilizzo delle risorse destinate alla realizzazione di specifici obiettivi del Piano sanitario nazionale, ai sensi dell’articolo 1, comma 34, della legge 23 dicembre 1996, n. 662.
Comma 401. A decorrere dal 1° gennaio 2017, nello stato di previsione del Ministero della Salute è istituito un Fondo per il concorso al rimborso alle regioni per l’acquisto dei medicinali oncologici innovativi, con una dotazione di 500 milioni di euro annui, mediante utilizzo delle risorse del comma 393.
Comma 406. La spesa per l’acquisto dei farmaci innovativi e dei farmaci oncologici innovativi concorre al raggiungimento del tetto della spesa farmaceutica per acquisti diretti di cui al comma 398 per l’ammontare eccedente annualmente l’importo di ciascuno dei fondi di cui ai commi 400 e 401.
Un approccio multidimensionale
Nella premessa dell’Allegato 1 della Determina n. 1535/2017, in cui vengono riportati i criteri per la valutazione dell’innovatività, l’AIFA specifica che per «l’attribuzione del carattere di innovatività di un farmaco in relazione a una specifica indicazione è necessario che venga dimostrato il valore terapeutico aggiunto, rispetto alle alternative terapeutiche disponibili, nel trattamento di una patologia grave, intesa come una malattia a esito potenzialmente mortale, oppure che induca ospedalizzazioni ripetute, o che ponga il paziente in pericolo di vita o che causi disabilità in grado di compromettere significativamente la qualità di vita».
Il modello di valutazione dell’innovatività, che al momento è unico per tutti i farmaci, ma che in futuro potrà prevedere l’utilizzo di indicatori specifici, prevede un approccio multidimensionale che tiene conto di tre elementi fondamentali:
il bisogno terapeutico;
il valore terapeutico aggiunto;
la qualità delle prove.
Bisogno terapeutico
Il bisogno terapeutico viene definito sulla base della disponibilità di alternative terapeutiche per la patologia in oggetto e degli esiti clinici ad esse associate. Ai fini del riconoscimento dell’innovatività in relazione a una specifica indicazione, il bisogno terapeutico, che è indice di quanto l’introduzione del nuovo trattamento sia necessario per far fronte alle esigenze terapeutiche di una popolazione di pazienti, viene classificato in cinque livelli – massimo, importante, moderato, scarso, assente (Tabella I) – descritti nel dettaglio nel Modulo per la richiesta del riconoscimento dell’innovatività (Allegato 2 della Determina n. 1535/2017).
Bisogno terapeutico
Massimo: assenza di opzioni terapeutiche per la specifica indicazione.
Importante: presenza di alternative terapeutiche per la specifica indicazione, ma che non producono impatto su esiti clinicamente rilevanti e validati per la patologia in oggetto.
Moderato: presenza di alternative terapeutiche per la specifica indicazione con impatto valutabile come limitato su esiti riconosciuti come clinicamente rilevanti e/o con un profilo di sicurezza incerto o non del tutto soddisfacente.
Scarso: presenza di una o più alternative terapeutiche per la specifica indicazione con impatto valutabile come elevato su esiti riconosciuti come clinicamente rilevanti e con un profilo di sicurezza favorevole.
Assente: presenza di alternative terapeutiche per la specifica indicazione in grado di modificare la storia naturale della malattia e con un profilo di sicurezza favorevole.
Valore terapeutico aggiunto
Massimo: maggiore efficacia dimostrata su esiti clinicamente rilevanti rispetto alle alternative terapeutiche (qualora disponibili). Il farmaco è in grado di guarire la malattia o comunque di modificarne significativamente la storia naturale.
Importante: maggiore efficacia dimostrata su esiti clinicamente rilevanti, o capacità di ridurre il rischio di complicazioni invalidanti o potenzialmente fatali, o migliore rapporto rischio/beneficio rispetto alle alternative, o capacità di evitare il ricorso a procedure cliniche ad alto rischio. Il farmaco modifica la storia naturale della malattia in una sottopopolazione di pazienti, o rappresenta comunque un vantaggio clinicamente rilevante, ad esempio in termini di qualità della vita e di intervallo libero dalla malattia, rispetto alle alternative terapeutiche disponibili.
Moderato: maggiore efficacia di entità moderata o dimostrata in alcune sottopopolazioni di pazienti o su esiti surrogati, e con effetti limitati sulla qualità della vita. Per condizioni nelle quali sia ammissibile l’assenza di un comparatore, disponibilità di evidenze suggestive di migliore efficacia clinica e profilo rischio/beneficio più favorevole rispetto alle alternative terapeutiche disponibili.
Scarso: maggiore efficacia che, tuttavia, è stata dimostrata su esiti non clinicamente rilevanti oppure risulta di scarsa entità. Vantaggi minori (ad esempio via di somministrazione più favorevole) rispetto alle alternative terapeutiche disponibili.
Assente: assenza di un beneficio clinico aggiuntivo rispetto alle alternative terapeutiche disponibili.
Qualità delle prove
Alta/moderata/bassa/molto bassa: valutazione secondo il metodo GRADE (Grading of Recommendations Assessment, Development and Evaluation)
Tabella I. Livelli per la classificazione del bisogno terapeutico, del valore terapeutico aggiunto e della qualità delle prove (Allegato 1 della Determina n. 1535/2017)
Box 2. Indicatori di efficacia in oncologia
Il gold standard è la sopravvivenza globale (OS).
In mancanza di dati di OS (che dovrà essere adeguatamente giustificata), in relazione al tipo di neoplasia e al setting terapeutico potranno essere considerati:
sopravvivenza libera da progressione (PFS);
sopravvivenza libera da malattia (DFS);
durata della risposta completa;
altri esiti surrogati di cui, anche in base all’entità dell’effetto, sia riconosciuto il valore predittivo di beneficio clinico.
L’adeguatezza dell’esito clinico selezionato sarà valutata anche in base al relativo profilo di tossicità.
Valore terapeutico aggiunto
Il valore terapeutico aggiunto viene definito sulla base dell’entità del beneficio clinico del nuovo trattamento, rispetto alle alternative terapeutiche disponibili, su esiti clinicamente rilevanti e validati per la patologia in oggetto. Nel Box 2 sono riportati gli indicatori di efficacia indicati dalla Determina per i farmaci oncologici. Anche in questo caso, ai fini del riconoscimento dell’innovatività il valore terapeutico aggiunto viene classificato in cinque livelli – massimo, importante, moderato, scarso, assente (Tabella I) – descritti nel dettaglio nel Modulo per la richiesta del riconoscimento dell’innovatività.
Qualità delle prove
Per la valutazione della qualità delle prove scientifiche portate a supporto della richiesta di innovatività AIFA ha deciso di adottare il metodo GRADE (Grading of Recommendations Assessment, Development and Evaluation) in base al quale la qualità verrà definita alta, moderata, bassa o molto bassa, come riportato nel Modulo per la richiesta del riconoscimento dell’innovatività.
Nella valutazione di farmaci indicati per le malattie rare, la Commissione Tecnico Scientifica (CTS) ha previsto che si possa attribuire l’innovatività anche con una qualità delle prove di livello basso, purché in presenza di un elevato bisogno terapeutico e di forti indicazioni di un beneficio terapeutico aggiunto.
L’iter di valutazione
La procedura per il riconoscimento del requisito di innovatività di un farmaco, rispetto a una specifica indicazione, ha inizio in seguito a sottomissione della richiesta da parte dell’Azienda titolare dell’AIC, o di specifica indicazione della CTS dell’AIFA in presenza di evidenze scientifiche (anche senza richiesta da parte dell’Azienda). Al termine dell’iter di valutazione la CTS formula il giudizio di innovatività in base al profilo derivante dalla combinazione delle valutazioni assegnate a bisogno terapeutico, valore terapeutico aggiunto e qualità delle prove. Di seguito vengono riportati i tre possibili esiti:
innovatività riconosciuta, in presenza di bisogno terapeutico e valore terapeutico aggiunto di livello massimo o importante e qualità delle prove alta. Per i farmaci con indicazione per le malattie rare (o altre malattie con tassi di prevalenza assimilabili), in presenza di un elevato bisogno terapeutico e di forti indicazioni di un beneficio terapeutico aggiunto, l’innovatività potrà essere riconosciuta anche se la qualità delle prove è bassa;
innovatività non riconosciuta, in presenza di un bisogno terapeutico e/o un valore terapeutico aggiunto di livello scarso o assente oppure qualità delle prove bassa o molto bassa;
situazioni intermedie che verranno valutate singolarmente e che possono portare al riconoscimento di innovatività condizionata (o potenziale).
Al termine del processo la CTS rende disponibile una breve relazione nella quale descrive le valutazioni relativi ai tre ambiti considerati ed esprime il giudizio finale. Nella Tabella II sono riportati i tre possibili esiti.
Permanenza del carattere di innovatività
Attribuzione dei benefici economici1
Accesso al fondo farmaci innovativi o farmaci oncologici innovativi
Inserimento nei Prontuari Terapeutici Regionali
Riconoscimento dell’innovatività
Massimo 36 mesi2
X
X
X
Riconoscimento dell’innovatività condizionata (o potenziale)
Massimo 18 mesi
–
–
X
Mancato riconoscimento dell’innovatività
–
–
–
–
Tabella II. Esiti della valutazione da parte della CTS e conseguenti benefici
1 Non applicazione degli sconti obbligatori (5% + 5%) e non applicazione del payback
2 Per il farmaco first in class, mentre eventuali followers che venissero in seguito riconosciuti innovativi in relazione alla medesima indicazione, la durata corrisponderà con il periodo residuo
Il richiedente potrà presentare controdeduzioni entro 10 giorni dalla comunicazione ricevuta dalla CTS, l’esito finale e la relazione verranno pubblicati sul sito web dell’AIFA contestualmente alla pubblicazione della determinazione di rimborsabilità e prezzo. Come riportato nelle prime righe dell’Allegato 1, infatti, l’ammissione alla rimborsabilità e il possibile riconoscimento dell’innovatività, pur basandosi sostanzialmente sulle stesse evidenze, rappresentano due procedure distinte tra le quali non esiste una consequenzialità automatica.
Procedura di rivalutazione
La Determina n. 1535/2017 prevede la possibilità di rivalutare un farmaco al quale è stata attribuita innovatività condizionata in seguito a istanza d’ufficio da parte di AIFA o richiesta dell’Azienda titolare di AIC. La procedura può essere attivata solo se sono disponibili nuove evidenze e dopo che sono decorsi almeno 18 mesi dalla prima valutazione. La rivalutazione può essere attivata anche per i farmaci ai quali è stata riconosciuta innovatività piena, nel caso emergano nuove evidenze. L’esito della rivalutazione può portare a mancata conferma del carattere di innovatività con conseguente rinegoziazione del prezzo e delle condizioni di rimborsabilità.
La procedura di riconoscimento di innovatività per una nuova indicazione di un farmaco che ha già ottenuto l’innovatività rispetto a un’altra indicazione potrà essere avviata dalla CTS autonomamente o su richiesta dell’azienda titolare dell’AIC.
Il bilancio del primo anno
Farmaci valutati
I farmaci ai quali viene riconosciuta innovatività piena e condizionata vengono inseriti negli elenchi dei farmaci innovativi dell’AIFA e pubblicati periodicamente sul sito dell’Agenzia. Attualmente, tra i farmaci non oncologici che accedono ai fondi emergono due macroaree terapeutiche: epatite C e malattie rare.
Da Aprile 2017 i farmaci che hanno ultimato la procedura di valutazione e per cui è terminato anche il processo di negoziazione del prezzo e definizione delle condizioni di rimborso sono 29. Di questi, a 11 è stata riconosciuta innovatività piena (di 5 oncologici), a 7 è stata attribuita innovatività condizionata, mentre ai restanti 11 non è stato riconosciuto carattere di innovatività (Tabella III).
Nome commerciale (principio attivo, azienda)
Indicazione
Bisogno terapeutico
Valore terapeutico aggiunto
Qualità delle prove
Innovatività riconosciuta
Alecensa (alectinib, Roche)
Carcinoma polmonare non a piccole cellule in stadio avanzato positivo per la chinasi del linfoma anaplastico (monoterapia, I linea)
Moderato
Importante
Moderata
Darzalex (daratumumab, Janssen Cilag)
Mieloma multiplo (in combinazione)
Moderato
Importante
Moderata
Dupixent (dupilumab, Sanofi-Aventis Groupe)
Dermatite atopica da moderata a grave in pazienti adulti eleggibili per la terapia sistemica
Profilassi della riattivazione e della malattia da citomegalovirus in pazienti adulti sieropositivi per CMV riceventi trapianto allogenico di cellule staminali ematopoietiche
Pazienti con storia clinica di neuroblastoma recidivante o refrattario, con o senza malattia residua
Massimo
Importante
Molto bassa
Rydapt (midostaurina, Novartis Europharm Limited)
Mastocitosi sistemica aggressiva, mastocitosi sistemica associata a neoplasie ematologiche o leucemia mastocitica (monoterapia)
Importante
Scarso
Bassa
Zalmoxis (linfociti T allogenici geneticamente modificati, MolMed)
Trapianto aploidentico di cellule staminali emopoietiche in neoplasie maligne ematologiche ad alto rischio
Moderato
Moderato
Molto bassa
Zavicefta (ceftazidime/avibactam, Pfizer)
Trattamento cIAI, cUTI e HAP, infezioni causate da microrganismi Gram-negativi aerobi
Importante
Scarso
Bassa e molto bassa
Zinplava (Bezlotoxumab, Merck Sharp & Dohme)
Prevenzione della recidiva dell’infezione da Clostridium difficile
Importante
Moderato
Bassa
Tabella III. Elenco dei farmaci valutati per l’innovatività dopo la pubblicazione della Determina n. 1535/2017 (ultimo aggiornamento 26/09/2018)
cIAI = infezione intra-addominale complicata; cUTI = infezione complicata del tratto urinario; HAP = polmonite acquisita in ospedale; HCV = virus dell’epatite C
Tra i farmaci che hanno ottenuto il riconoscimento di innovatività piena, due sono indicati nel trattamento dell’HCV (Maviret e Vosevi) ed entrambi hanno ricevuto giudizio “importante” alle voci bisogno terapeutico e valore terapeutico aggiunto e giudizio “moderato” per quanto riguarda la qualità delle prove. In questo caso, anche in assenza di qualità delle prove alta, è stata loro riconosciuta l’innovatività in quanto erano presenti le stesse caratteristiche che hanno consentito di riconoscere l’innovatività agli altri farmaci della stessa classe. I benefici riconosciuti dureranno fino alla decadenza dell’innovatività di Epclusa (first in class). Due sono i farmaci oncologici ad aver ottenuto l’innovatività dopo l’emanazione della Determina n. 1535/2017: Tecentriq, in relazione all’indicazione per il trattamento in monoterapia del carcinoma polmonare non a piccole cellule localmente avanzato o metastatico, e Darzalex in relazione all’indicazione per il trattamento del mieloma multiplo in combinazione con lenalidomide e desametasone (o bortezomib e desametasone). In quest’ultimo caso, anche se il bisogno terapeutico è stato giudicato “moderato”, il valore terapeutico aggiunto “importante” e la qualità delle prove “moderata”, a Darzalex è stata riconosciuta l’innovatività, mentre in relazione all’indicazione per il trattamento del mieloma multiplo in monoterapia, pur in presenza di un bisogno clinico “importante”, non gli è stata riconosciuta l’innovatività a causa di un valore terapeutico aggiunto di difficile definizione e una qualità delle prove giudicata “bassa”.
Una qualità delle prove “bassa” è stata attribuita anche a Oxervate rispetto all’indicazione nel trattamento della cheratite neurotrofica, tuttavia trattandosi di un farmaco orfano, e in presenza di un bisogno terapeutico e di un valore terapeutico aggiunto valutati come “importanti”, gli è stata riconosciuta l’innovatività. Infine, anche Spinraza ha ricevuto l’innovatività, nonostante la qualità delle prove sia stata giudicata bassa, in virtù della rarità della patologia (atrofia muscolare spinale 5q) e dell’assenza di alternative terapeutiche (il bisogno terapeutico è stato infatti definito “massimo”).
In generale, dall’analisi delle schede di valutazione pubblicate dall’AIFA, il parametro più influente sembra essere il valore terapeutico aggiunto: tra i farmaci con innovatività riconosciuta e condizionata infatti il valore terapeutico aggiunto è sempre “importante” o “moderato”. In presenza di un valore terapeutico aggiunto “scarso”, invece, anche in presenza di un bisogno terapeutico “importante”, non è stata riconosciuta l’innovatività (Tabella III).
Fondi spesi
Dai dati riportati nel Monitoraggio AIFA della Spesa Farmaceutica Nazionale e Regionale relativo al periodo gennaio-dicembre 2017, è emerso che le Regioni hanno speso quasi 144 milioni di euro per l’acquisto dei farmaci innovativi non oncologici e poco più di 400 milioni per i farmaci innovativi oncologici, per un totale di circa 550 milioni, pari a poco più del valore di un singolo fondo (cfr. Appendice). Una delle ragioni della mancata spesa dei fondi erogati potrebbe essere legata al ritardo con il quale è stato pubblicato il Decreto che ha definito le modalità operative di erogazione dei fondi, la cui assenza potrebbe aver creato incertezza nelle Regioni nel corso del 2017. Inoltre il risultato del 2017 potrebbe anche essere stato influenzato dal computo del payback per i farmaci anti-epatite C relativo agli anni 2016 e 2017.
Nel 2017 le Regioni hanno speso quasi 144 milioni di euro per l’acquisto dei farmaci innovativi non oncologici e poco più di 400 milioni per i farmaci innovativi oncologici
Il Decreto del 16 febbraio 2018 ha sostituito il Decreto del 21 dicembre 2017 e ha stabilito che le Regioni, attraverso un sistema a rimborso (acconto attraverso il criterio del riparto del Fondo Sanitario Nazionale e un conguaglio a valle), provvedano alla ripartizione delle risorse stanziate. Secondo l’ultimo rapporto OsMed, nonostante una relativamente bassa spesa per l’acquisto di farmaci innovativi non oncologici, nel 2017 i consumi sono aumentati di quasi il 12% rispetto al 2016 permettendo un più ampio accesso alle terapie innovative. La riduzione della spesa registrata, pari a circa il 38% rispetto al 2016, sembrerebbe essere correlata alla negoziazione dei prezzi avvenuta nel corso del 2017, soprattutto per i farmaci per il trattamento dell’HCV.
I dati provvisori riportati nel documento di Monitoraggio AIFA della Spesa Farmaceutica Nazionale e Regionale relativo al periodo gennaio-aprile 2018 riportano cifre in merito all’utilizzo dei fondi con una spesa pari a quasi 284 milioni per i farmaci innovativi non oncologici (non sono presenti tetti di prodotto e accordo prezzo/volume) e poco più di 177 milioni per i farmaci innovativi oncologici.
Ai fini di una migliore allocazione delle risorse e per valutare la congruità dei fondi stanziati per i farmaci innovativi la Legge di Bilancio 2018, nei commi 408 e 409 dell’articolo 1, stabilisce l’avvio di «un monitoraggio degli effetti dell’utilizzo dei farmaci innovativi e innovativi oncologici sul costo del percorso terapeutico-assistenziale complessivo». Il monitoraggio verrà effettuato su una o più aree terapeutiche e si baserà sui dati di Real World Evidence e sulle informazioni riportate nei registri di farmaci innovativi sottoposti a monitoraggio AIFA.
Bibliografia di riferimento
AIFA. Determina n. 1535/2017. Gazzetta Ufficiale n. 218 del 18-09-2017
L’uso dei farmaci in Italia – Rapporto OsMed 2017.
Legge 11 dicembre 2016, n. 232. Gazzetta Ufficiale n. 297 del 21-12-2016
Legge 27 dicembre 2017, n. 205. Gazzetta Ufficiale n. 302 del 29-12-2017
Ministero della Salute. Decreto 16 febbraio 2018 . Gazzetta Ufficiale n. 81 del 07-04-2018
Appendice
Regione
A
(B = A / Totale) × 100
C=A2
Spesa tracciabilità (spesa totale)
Inc. % spesa tracciabilità
Spesa al netto dei PayBack
Piemonte
27.573.168
6,70
27.573.168
Valle d’Aosta
567.602
0,10
567.602
Lombardia
57.677.432
14,10
57.677.432
P.A. Bolzano
4.142.696
1,00
4.142.696
P.A. Trento
2.746.301
0,70
2.746.301
Veneto
26.960.378
6,60
26.960.378
Friuli VG
11.073.687
2,70
11.073.687
Liguria
13.520.511
3,30
13.520.511
Emilia Romagna
37.166.614
9,10
37.166.614
Toscana
34.642.607
8,50
34.642.607
Umbria
7.066.417
1,70
7.066.417
Marche
12.697.864
3,10
12.697.864
Lazio
42.156.394
10,30
42.156.394
Abruzzo
9.096.320
2,20
9.096.320
Molise
1.741.361
0,40
1.741.361
Campania
42.206.925
10,30
42.206.925
Puglia
29.074.904
7,10
29.074.904
Basilicata
3.746.988
0,90
3.746.988
Calabria
9.117.554
2,20
9.117.554
Sicilia
24.880.980
6,10
24.880.980
Sardegna
11.389.689
2,80
11.389.689
Italia
409.246.391
100,00
409.246.391
Spesa farmaceutica gennaio-dicembre 2017 per i medicinali innovativi1 oncologici che accedono al fondo del Ministero della Salute, previsto ai sensi dell’art. 1, commi 402, 403 e 404, della legge 11 dicembre 2016, n. 232, legge di Bilancio 2017 – (Dato Tracciabilità DM 2004). Modificata da Monitoraggio della Spesa Farmaceutica Nazionale e Regionale gennaio-dicembre 2017 (aggiornamento 01/06/2018)
1 L’elenco di medicinali che alla data del 31 dicembre 2017 accedono al fondo degli innovativi oncologici include: Abraxane, Imbruvica, Keytruda, Opdivo, Perjeta, Zydelig. Perjeta ha perso il requisito di innovatività in data 7 Luglio 2017. Per il farmaco Abraxane si considera il valore complessivo che comprende oltre ai flussi per l’indicazione terapeutica “adenocarcinoma metastatico del pancreas”, per cui il farmaco possiede il requisito dell’innovatività, anche i dati relativi alle prescrizioni per “tumore metastatico della mammella”, attualmente non scorporabili dal totale.
2 Per il periodo gennaio–dicembre non risultano Payback relativi ai farmaci innovativi oncologici.
Regione
A
(B = A / Totale) × 100
C
D = A-C
Spesa tracciabilità2
(spesa totale)
Inc. % spesa tracciabilità
Payback innovativi non oncologici Classe A3
Spesa al netto dei PayBack
Piemonte
67.756.389
7,50
55.527.840
12.228.549
Valle d’Aosta
957.368
0,10
843.928
113.441
Lombardia
175.049.311
19,40
124.036.017
51.013.294
P.A. Bolzano
3.303.993
0
2.441.599
862.394
P.A. Trento
3.620.706
0,40
3.077.357
543.349
Veneto
58.684.975
6,50
45.412.999
13.271.976
Friuli VG
12.706.382
1,40
7.675.373
5.031.010
Liguria
23.905.708
2,70
19.239.492
4.666.216
Emilia Romagna
64.462.014
7,20
58.196.267
6.265.748
Toscana4
47.667.935
5,30
49.791.012
-2.123.077
Umbria
10.111.451
1,10
6.564.791
3.546.660
Marche
14.831.723
1,60
12.358.591
2.473.132
Lazio
68.262.322
7,60
58.862.486
9.399.837
Abruzzo
12.934.109
1,40
9.383.831
3.550.278
Molise
3.497.244
0,40
3.403.203
94.041
Campania
131.128.001
14,60
126.537.300
4.590.701
Puglia
66.511.052
7,40
61.248.702
5.262.350
Basilicata
8.317.386
0,90
6.219.793
2.097.593
Calabria
30.860.818
3,40
26.827.329
4.033.489
Sicilia
67.116.723
7,50
54.542.899
12.573.824
Sardegna
28.675.291
3,20
24.457.885
4.217.407
Italia
900.360.903
100,00
756.648.693
143.712.210
Spesa farmaceutica gennaio-dicembre 2017 per i medicinali innovativi1 non oncologici che accedono al fondo del Ministero della Salute, previsto ai sensi dell’art. 1, commi 402, 403 e 404, della legge 11 dicembre 2016, n. 232 , legge di Bilancio 2017 – (Dato Tracciabilità DM 2004). Modificata da Monitoraggio della Spesa Farmaceutica Nazionale e Regionale gennaio-dicembre 2017 (aggiornamento 01/06/2018)
1 L’elenco di medicinali che alla data del 31 dicembre 2017 accedono al fondo degli innovativi non oncologici include: Daklinza, Exviera, Harvoni, Kalydeco, Olysio, Sovaldi, Viekirax, Zepatier, Epclusa, Spinraza e Maviret. Nell’elenco è compreso inoltre il farmaco Strimvelis, di classe H, per cui non sono presenti dati di tracciabilità. Con Determina AIFA del 22 maggio 2017 (G. U. n.126 del 01/06/2017) le specialità Sovaldi e Harvoni a partire dal 02/06/2017 sono riclassificate in fascia C , pertanto dalla data dal 2 giugno 2017 non contribuiscono più alla spesa farmaceutica del SSN e sono esclusi dal fondo degli innovativi.
2 I dati di tracciabilità per il farmaco Epclusa sono al netto delle note di credito relative all’accordo p/v vigente. Per il farmaco Olysio non sono presenti dati di tracciabilità per i mesi di luglio, agosto, settembre e dicembre (AIC 043441017) e per il mese di Agosto (AIC 043441029).
3 Comprende: i) tetti di prodotto e Accordi Prezzo /Volume gennaio-dicembre 2017 per un totale di 306,7 milioni di euro. Sono compresi i payback pubblicati in GU nel periodo indicato con dettaglio regionale (seguendo il criterio di cassa) relativi al farmaco Kalydeco per un valore pari a 3,1 milioni (GU 34 del 10.2.2017), e per i farmaci Sovaldi e Harvoni i PayBack previsti dalla Determina AIFA del 12 aprile 2017 e pubblicati in GU 94 dl 22,4,2017 (rispettivamente 28,7 e 36,0 milioni di euro), i PayBack pubblicati in GU 159 del 10-07-2017 ( rispettivamente 106,8 e 109,4 milioni di euro) e i PayBack pubblicati in GU 297 del 21.12.2017 (rispettivamente 13,1 e 9,7 milioni); ii) MEA per un valore pari 449,9 milioni di euro. Con riferimento ai MEA , l’AIFA ha avviato una attività di verifica e validazione dei dati relativamente all’Importo effettivamente pagato dalle Aziende rispetto alle PDP (Proposte di Pagamento), utilizzate per il calcolo dell’Importo nei precedenti monitoraggi. Il dato è oggi disponibile per tutte le aziende interessate e si riferisce ai dati estratti dalla piattaforma al 25.05.2018 per l’anno 2017. Essendo il registro uno strumento dinamico, i valori dei MEA possono risultare diversi rispetto a precedenti estrazioni relative al medesimo periodo di osservazione.
4 Il dato della spesa netta per la regione Toscana risulta negativo per effetto di valori dei PB più elevati rispetto ai dati di spesa di tracciabilità. Tali valori sono in particolare determinati di PB relativi ai farmaci Sovaldi e Harvoni con competenza precedente all’anno 2017.
Quando si affronta il tema dei farmaci innovativi, anche tra stakeholders, a mio parere il primo elemento da sottolineare è la necessità di una terminologia comune e accurata, per definire in modo univoco e senza rischio di fraintendimenti che cosa si intenda con la denominazione “farmaco innovativo”. Come spesso avviene in Italia, si rischia infatti di fare confusione, con i diversi interlocutori che utilizzano lo stesso termine per indicare elementi diversi.
Vorrei pertanto procedere per esclusione e liberare il campo dai possibili equivoci: esistono molteplici farmaci che potrebbero essere definiti come innovativi. Ad esempio ci sono i medicinali “game-changing”, che cambiano radicalmente il sistema di riferimento, oppure quelli che modificano in maniera significativa il decorso della patologia e la vita futura dei pazienti, oppure quelli che presentano caratteristiche originali rispetto ai precedenti (una nuova classe, un nuovo meccanismo d’azione, una tecnologia o un device completamente nuovi), oppure semplicemente tutti i farmaci nuovi, che in quanto lanciati sul mercato sicuramente vanno a coprire un bisogno di salute, piccolo o grande che sia, che prima non era evaso. Tutti questi medicinali comportano sicuramente una serie di vantaggi fondamentali per il paziente e per il sistema, e anche quelli che per definizione non apportano vantaggi diretti in termini di salute (es. generici, biosimilari, me-too), hanno vantaggi indiretti creando competizione. Seppure tutti questi farmaci possano essere definiti innovativi, la legge di Bilancio del 2017 ci fornisce una nuova definizione: saranno innovativi i farmaci che accederanno ai fondi per i farmaci oncologici e per i farmaci non oncologici. Tale attribuzione, meramente economica, segue però criteri stabiliti dall’Agenzia Italiana del Farmaco tramite la Commissione Tecnico Scientifica (CTS).
La definizione di “innovativo” non rientra tra le priorità del paziente che, in quanto tale, vuole essere curato al meglio ma non è interessato all’innovatività o meno della cura in sé. Può rientrare invece tra le priorità del Servizio Sanitario Nazionale nella misura in cui consente un Early Access, cioè consente al farmaco di essere messo a disposizione del paziente in tempi rapidi, per poter offrire una soluzione che prima non c’era. Inoltre potrebbe essere utile al sistema in generale poiché permette di individuare le priorità espresse dal SSN e può consentire alle aziende farmaceutiche di orientare il proprio piano di ricerca & sviluppo secondo quelle direttrici. Infine, l’istituzione dei fondi per i farmaci innovativi ha rappresentato un vantaggio in termini di extra-budget per la spesa farmaceutica, anche nell’ottica di gestire una contingenza in attesa di nuove regole, di una nuova governance.
Come noto, la valutazione della CTS si basa su tre criteri: il bisogno terapeutico, il valore terapeutico aggiunto e la qualità delle prove. Per bisogno terapeutico intendiamo qual è il contesto nel quale si situa il farmaco in analisi: ad esempio, quanto è grave la patologia, quale priorità riveste all’interno del Servizio Sanitario Nazionale, quali altre opzioni terapeutiche sono già disponibili e qual è il loro livello di efficacia, come vengono gestiti globalmente i pazienti affetti da quella determinata patologia. Con il parametro del valore terapeutico aggiunto si valutano i vantaggi aggiuntivi che il farmaco in studio è in grado di apportare rispetto ai comparator evidenziati nell’analisi del bisogno terapeutico, in termini sia di rischi sia di benefici. Infine, la Commissione esamina la qualità delle prove per determinare la certezza o l’incertezza del valore terapeutico aggiunto rilevato. La qualità delle prove è determinante per definire la fiducia con la quale si può attribuire il valore terapeutico aggiunto: in mancanza di evidenze robuste a supporto del farmaco candidato, l’innovatività non può essere concessa. Parziale eccezione deve farsi in caso di malattie rare che, per definizione, possono contare su un minor numero di studi e casi clinici. Tra questi tre criteri, il valore terapeutico aggiunto può essere considerato come il “driver” del processo, cioè il fattore che pesa maggiormente nell’attribuzione del carattere di innovatività.
Al termine dell’iter di valutazione, la normativa prevede una serie di vantaggi concessi al farmaco riconosciuto come innovativo nella sola indicazione per la quale è stata avanzata la richiesta, e che dovrebbero, nelle intenzioni del legislatore, favorirne l’accesso ai pazienti: non partecipa al payback, non è soggetto allo sconto obbligatorio del 5%+5% ed è immediatamente inserito nei Prontuari Terapeutici Regionali. I vantaggi economici non sono concessi ai farmaci ai quali si riconosce una innovatività condizionata, della durata di 18 mesi, da rivalutare, mentre l’accesso immediato ai Prontuari Terapeutici Regionali è comunque assegnato.
A mio parere, la definizione dei criteri per l’innovatività e il processo intrapreso presentano dei punti di forza e dei punti di debolezza. Innanzitutto un punto di forza fondamentale è la trasparenza dell’iter valutativo e dei risultati, che sono stati resi pubblici e messi a disposizione di tutti gli interessati sul sito dell’AIFA: gli assessment sono consultabili liberamente e le eventuali incoerenze, che in Commissione abbiamo cercato di evitare, potrebbero essere evidenziate e discusse, anche in un’ottica di miglioramento per il futuro. Più avanti, in questa rivista, si troverà una tabella dei farmaci valutati con gli esiti, un processo di trasparenza che ben fa sperare per il futuro.
Inoltre mi preme di sottolineare come i tre criteri individuati (bisogno terapeutico, valore terapeutico aggiunto e qualità delle prove) siano, nella sostanza, coerenti e condivisi con quanto predisposto da altri organismi regolatori internazionali, sia a livello europeo, sia nell’America del Nord, sia in Estremo Oriente.
Infine ritengo che questi criteri siano utili anche perché definiscono l’ambito in cui si muove il Servizio Sanitario Nazionale che riconosce come prioritari i farmaci per cui esiste un alto bisogno terapeutico, con un valore terapeutico aggiunto importante e che possano contare su prove sufficientemente robuste per corroborarlo.
Non possiamo però mancare di menzionare anche alcuni punti di debolezza, ascrivibili non solo ai criteri ma all’intero percorso.
Il primo aspetto che vorrei evidenziare è che l’inserimento nell’elenco dei farmaci innovativi dura 3 anni e, secondo me, questo rappresenta un periodo troppo lungo. È una scelta, e forse si potrebbe valutare se riconsiderarla. Poiché altri problemi affliggono il sistema, e uno di questi è rappresentato dalle storture che provoca il budget di prodotto per le Aziende nell’anno di lancio, si potrebbe immaginare che immettere più farmaci per un periodo più breve potrebbe essere di aiuto. Inoltre, un budget triennale implica una certa capacità del sistema a prevedere le spese future, e questo potrebbe essere tra i motivi per i quali non tutti i fondi per i farmaci innovativi sono stati spesi nel 2017.
Il secondo aspetto critico chiama in causa il rischio che, con l’attribuzione del carattere di innovatività e i conseguenti benefici, anche economici, si possa premiare un farmaco che, probabilmente, verrebbe già premiato in un’altra maniera: ad esempio perché, essendo game-changing, con un impatto molto importante sulla gestione di una patologia e dei vantaggi in grado di cambiare sostanzialmente la vita dei pazienti, avrebbe ugualmente delle quote di mercato molto alte, riuscirebbe a negoziare un prezzo molto alto in Comitato Prezzi e Rimborso (CPR), e avrebbe un accesso regionale privilegiato. Questa “concentrazione” di vantaggi sugli stessi farmaci potrebbe assumere un ruolo negativo e creare addirittura delle distorsioni del mercato.
Un altro aspetto riguarda l’eventualità che la normativa sui fondi innovativi e il relativo processo di valutazione vadano a sovrapporsi ad altre norme già esistenti in ambito farmaceutico, come ad esempio il fondo nazionale per l’impiego di farmaci orfani per le malattie rare. Poiché anche i farmaci rari non partecipano al ripiano della spesa farmaceutica tramite payback, ha senso che un farmaco raro venga inserito nell’elenco dei farmaci innovativi o assistiamo ad un sovrapporsi di procedure e valutazioni? Anche in questo ambito, mi sembrerebbe opportuno un lavoro di riordino e revisione del sistema.
Un ultimo elemento critico che vorrei evidenziare e che nasce come riflessione dall’esperienza in CTS è che uno degli obiettivi dell’istituzione dei fondi per i farmaci innovativi era velocizzare l’accessibilità ai pazienti per alcuni farmaci giudicati particolarmente importanti: in questo senso, non penso che l’obiettivo sia stato raggiunto pienamente in quanto, anche in presenza del riconoscimento di innovatività, la procedura per l’immissione in commercio rimane quella standard, esattamente come avverrebbe per qualsiasi altro medicinale. A livello regionale penso che in alcuni casi si sia ottenuto effettivamente un Early Access per il farmaco innovativo, ma a livello nazionale questo non è avvenuto. Un sistema simile alla francese Autorisation Temporaire d’Utilisation (ATU) per i farmaci definiti innovativi attraverso i criteri AIFA potrebbe rappresentare un ulteriore valore aggiunto. La possibilità di garantire un effettivo Early Access ai farmaci innovativi è un tema che, secondo me, merita di essere affrontato perché ci sono ancora ampi margini di miglioramento per arrivare ai pazienti in tempi brevi.
Per concludere, vorrei presentare anche una provocazione personale sul tema: avendo individuato tre criteri che ci aiutano a definire quanto ci serve un farmaco (bisogno terapeutico), quanto è più efficace rispetto a quello che abbiamo già a disposizione (valore terapeutico aggiunto) e quanto siamo sicuri di questa nuova opportunità (qualità delle prove), perché non utilizzare questi stessi criteri per l’intera procedura di prezzo & rimborso? A mio parere, qualunque farmaco potrebbe essere valutato in questo modo e la CPR, le aziende e tutto il SSN potrebbero lavorare meglio, in presenza di trasparenza delle valutazioni.
Fonte
Il presente editoriale è tratto dal Convegno “L’adozione di farmaci innovativi nella pratica clinica”, promosso dall’Accademia Nazionale di Medicina (Bologna, 4-5 giugno 2018)








 nella pratica? Gli esempi da alcune Regioni")
 Come viene gestito il fondo farmaci innovativi nella sua Regione e come viene ripartito tra le diverse aziende sanitarie?
Come viene gestito il fondo farmaci innovativi nella sua Regione e come viene ripartito tra le diverse aziende sanitarie? Come viene gestito l’accesso al fondo farmaci innovativi nella sua Regione e la ripartizione tra le diverse aziende sanitarie?
Come viene gestito l’accesso al fondo farmaci innovativi nella sua Regione e la ripartizione tra le diverse aziende sanitarie? Come viene gestito l’accesso al fondo dei farmaci innovativi nella sua Regione e la ripartizione tra le diverse aziende sanitarie?
Come viene gestito l’accesso al fondo dei farmaci innovativi nella sua Regione e la ripartizione tra le diverse aziende sanitarie? Come viene gestito il fondo farmaci innovativi nella sua Regione e la ripartizione tra le diverse aziende sanitarie? Quali sono le direttive regionali?
Come viene gestito il fondo farmaci innovativi nella sua Regione e la ripartizione tra le diverse aziende sanitarie? Quali sono le direttive regionali?