Alcuni (dis)orientamenti giurisprudenziali recenti
L’importanza del settore dei farmaci biologici spiega perché, nonostante molti anni siano passati dall’introduzione dei primi farmaci biosimilari e dopo che faticosamente, anche per l’assenza di norme specifiche, la giurisprudenza abbia trovato punti di equilibrio funzionali e funzionanti per il sistema, i casi giudiziali che coinvolgono il rapporto tra originatori e biosimilari siano sempre molto presenti nella giurisprudenza amministrativa.
Talvolta, tuttavia, si ha l’impressione che ci si dimentichi dell’intero percorso evolutivo che la giurisprudenza, e la legge sulla scorta di essa, hanno compiuto negli ultimi dieci anni. Succede cioè che l’attenzione del giudice si concentri su aspetti di dettaglio della biosimilarità che, seppur effettivamente importanti e meritevoli di approfondimento individuale, tuttavia non possono essere esaminati senza tenere in considerazione l’intero quadro normativo, scientifico e – anche – giurisprudenziale in questa materia.
In particolare, due recenti decisioni dimostrano come in alcuni casi si finisca facilmente per complicare situazioni che, al contrario, dovrebbero ormai essere molto semplici.
Il caso che ha portato alla decisione del T.A.R. per la Toscana, Sez. II, n. 400 del 21 marzo 2019
Notevole è stato il risalto mediatico che la recente sentenza del T.A.R. per la Toscana, sez. II, n. 400 del 21 marzo 2019, ha avuto sulla stampa specializzata, che l’ha perlopiù enfatizzata per le posizioni di chiusura rispetto al biosimilare rispetto all’originatore (mi riferisco specialmente all’articolo apparso sul quotidiano Sanità24 del 26 marzo 2019, nel quale peraltro è riportato un numero della sentenza errato, dall’eloquente titolo “Biologici o biosimilari, sui farmaci va salvaguardata l’autonomia del medico”).
Il T.A.R. di Firenze, in effetti, non ha affatto chiuso ai biosimilari né affermato che tra biosimilare ed originatore il medico è libero di decidere in completa autonomia. Questa posizione sarebbe semplicemente in contrasto con le norme e con i principi giurisprudenziali tuttora in vigore.
Metodologicamente, l’esame della sentenza in questione non può prescindere dal punto di partenza essenziale: il contenuto del provvedimento impugnato. E qui deve essere rimarcato come, sorprendentemente, la delibera della Regione Toscana n. 194 del 2018 aveva previsto modalità di prescrizione, dispensazione e acquisizione di farmaci senza distinguere tra farmaci chimici (e relativi generici) e farmaci biotecnologici (e relativi biosimilari). Si erano previste, cioè, regole amministrative uniche, senza prevedere alcuna specificità per i farmaci biotecnologici. E correttamente il T.A.R. dà atto che questa è la premessa del proprio ragionamento, quando riconosce che la delibera impugnata è riferita ai farmaci in generale e non contiene alcuna regolamentazione specifica sui farmaci biologici e che, di conseguenza, risulta applicabile anche a questi ultimi.
In particolare, l’ente regionale aveva stabilito una procedura di controllo, da parte della Regione stessa, su eventuali richieste di acquisto di farmaci diversi da quelli aggiudicati inserite nel sistema informativo regionale, e che tale richiesta sarebbe stata esaminata in base, esclusivamente, alle politiche di governance della spesa e della sostenibilità dell’assistenza farmaceutica regionale.
Di fronte ad un contenuto di questo tipo è francamente inimmaginabile un esito diverso da quello dell’annullamento. Infatti, l’errore commesso dalla Regione è evidente sotto un duplice profilo: a) non avere previsto regole specifiche per i farmaci biologici; b) non avere tenuto conto che nel rapporto tra originator e biosimilari vi sono una serie di norme (art. 15 d.l. n. 95/2012 nel testo oggi vigente) e di regole (il secondo position paper di AIFA, in particolare) che prevedono un controllo medico sulla prescrizione.
È del tutto condivisibile, in questa ottica, che il T.A.R. per la Toscana abbia rimarcato alcuni principi imprescindibili quando si ha a che fare con i biosimilari.
Primo: ai farmaci biologici non si applica l’art. 7 d.l. n. 347/01 conv. in l. n. 405/01 che consente, per i soli farmaci chimici, la c.d. sostituibilità secondaria, ossia quella automatica, operata dal farmacista che fornisce al paziente il farmaco generico in assenza di specifica indicazione del medico prescrittore circa la non sostituibilità del farmaco prescritto. Per i biosimilari, tale sostituibilità secondaria, o automatica, è esclusa dall’art. 15, comma 11 quater, d.l. n. 95/12 conv. in l. n. 135/2012 ed introdotto dalla legge n. 232/16 che prevede sempre l’esclusivo controllo del medico sulla prescrizione.
Secondo: per i farmaci biologici non è consentito effettuare una gara ponendo nel medesimo lotto principi attivi differenti quando vi siano anche biosimilari (art. 15, comma 11 quater, comma 3, primo periodo, d.l. n. 95/12).
Terzo: le procedure di acquisto – cui anche la delibera regionale si riferiva – sono diverse, dal momento che quando i biosimilari siano almeno tre è obbligatoria la procedura di accordo quadro con tre aggiudicatari (art. 15, comma 11 quater, comma 3, lett. b).
Quarto: il medico che prescriva un farmaco biologico aggiudicato con questa particolare forma di procedura obbligatoria di accordo quadro con più operatori è libero di prescrivere uno dei primi tre farmaci, se ciò è preordinato a garantire la continuità terapeutica del paziente.
Libertà prescrittiva e governance della spesa sono due facce della stessa medaglia, non elementi contrapposti
La sentenza avrebbe potuto concludersi qui: questi quattro punti mostrano l’illegittimità del provvedimento della Regione, per non avere previsto queste peculiarità – peraltro discendenti direttamente dalla legge – per i farmaci biologici e biosimilari.
Ma è in questo contesto che va valutata l’ulteriore affermazione, quasi fosse un obiter dictum nella sentenza, sulla quale successivamente si sono concentrati tutti i commenti pro-originatore. Il Collegio infatti aggiunge che “se è ben possibile che la Regione eserciti, nell’ambito di politiche di razionalizzazione della spesa farmaceutica, una funzione di orientamento nei confronti dei medici prescrittori, permane tuttavia un “limite invalicabile” che è dato dall’autonomia decisionale del medico nella prescrizione di un farmaco, sotto il profilo dell’appropriatezza terapeutica”.
Il rapporto tra costo, costo-beneficio e libertà prescrittiva è infatti da tempo solcato dalla giurisprudenza amministrativa, che pare in effetti avere trovato un punto di equilibrio convincente. Il Consiglio di Stato ha da qualche anno precisato che “costituisce dovere, anche per il medico prescrittore quello di scegliere, tra prodotti ritenuti di pari efficacia, quello meno oneroso per il servizio sanitario nazionale” (Sez. III, sent. n. 5705 del 17 dicembre 2015 e n. 5776 del 18 dicembre 2015). Il concetto è stato ribadito anche successivamente (Sez. III, sent. n. 3621 del 21 luglio 2017) proprio in riferimento a provvedimenti regionali, valutati legittimi da Palazzo Spada, che prevedevano conseguenze a carico dei medici prescrittori che non tenessero in considerazione, all’atto della prescrizione, anche le valutazioni di costo della terapia unitamente alle esigenze terapeutiche, motivando specificamente perché intendessero discostarsi dalla prescrizione del farmaco a minor costo (per intolleranze, per continuità terapeutica, ecc.).
La più recente posizione del Consiglio di Stato, citata dalla stessa sentenza toscana, ha ulteriormente meglio precisato i rapporti esistenti tra le esigenze di contenimento della spesa farmaceutica regionale e la libertà prescrittiva del medico, ritenendo legittima e valorizzando “l’espressa previsione (…) che – qualora il medico ritenesse di non poter utilizzare il farmaco biosimilare o biologico originator al costo di terapia più basso rispetto al costo di terapia di altro farmaco biosimilare o biologico originator – fosse tenuto a motivare la scelta terapeutica, con specifica relazione indirizzata alla direzione sanitaria di appartenenza e di competenza territoriale dell’assistito” (Sez. III, n. 2821 del 11 maggio 2018). Cosicché, qualora tali cautele manchino, imponendo per esempio ai medici una rigida soglia minima di prescrizione di farmaco biosimilare o comunque a minor costo, i provvedimenti sarebbero al contrario illegittimi per violazione della libertà prescrittiva del medico.
Ecco, allora, che anche la sentenza del T.A.R. per la Toscana, pur nelle conclusioni del tutto corretta e condivisibile, appare forse un po’ troppo superficiale nell’affermare che la libertà prescrittiva è “limite invalicabile” e che le politiche di governance della spesa farmaceutica regionale non avrebbero alcun rilievo nella “valutazione medica di appropriatezza di cura e di garanzia della continuità terapeutica”.
La libertà prescrittiva deve essere garantita, certo: ma non è arbitrio prescrittivo. Deve essere esercitata dal medico all’interno del quadro di regole e di norme esistenti, tra cui l’indicazione (giurisprudenziale ma anche contenuta nell’art. 13 del codice di deontologia medica) secondo cui il costo della terapia è uno degli elementi che concorre, assieme alle valutazioni terapeutiche e cliniche, nella scelta del farmaco da prescrivere. Con la conseguenza che, a parità di effetti terapeutici tra biosimilare ed originator va preferito il farmaco che costa meno. Occorre solo precisare che questa parità di effetti va valutata nel caso concreto, ossia sul singolo paziente da parte del singolo medico; ciò dal momento che ex ante ed in astratto la parità di effetti terapeutici è ontologicamente insita nel concetto e nella definizione di farmaco biosimilare, nel quale le differenze con l’originator, pur esistenti ed anzi ineliminabili, sono già state valutate da EMA all’atto dell’autorizzazione all’immissione in commercio come non significative ai fini della conclusione che i due farmaci hanno lo stesso profilo di efficacia, sicurezza e d’uso.
Volendo sintetizzare: spesa farmaceutica e appropriatezza sono due facce della stessa medaglia. La prima non può prevalere sulla seconda. Ma neppure il contrario.
Lo strano caso dell’epoetina zeta
In questo contesto, di apparente scarsa attenzione sul valore scientifico del concetto di biosimilarità, pare interessante commentare brevemente anche la decisione, questa sì scarsamente condivisibile anche nelle conclusioni, con la quale il Consiglio di Stato ha ritenuto che l’epoetina zeta non sia un biosimilare dell’epoetina alfa (si tratta della sentenza della Sez. III, n. 871 del 5 febbraio 2019).
La sentenza infatti muove correttamente dall’esame dell’autorizzazione EMA dell’epoetina zeta, riconoscendo che effettivamente il farmaco è autorizzato come biosimilare e che il medicinale di riferimento è l’epoetina alfa (originatore, ovviamente). Tanto basterebbe per rispondere al quesito giuridico in senso diametralmente opposto rispetto a quanto affermato dalla sentenza stessa.
Sennonché, il ragionamento del Collegio si è concentrato su di una relazione istruttoria, acquisita al fascicolo, commissionata in primo grado con la quale era stato chiesto ad AIFA di chiarire cosa fosse l’epoetina zeta.
Ed in quella relazione, inevitabilmente, AIFA ha spiegato che il concetto di farmaco biosimilare ammette, ed anzi – aggiungo – non può prescindere, dal riconoscimento delle diversità esistenti rispetto al farmaco originator. Ciò è vero per qualsiasi tipo di farmaco biologico, ed è vero peraltro anche tra lotti produttivi diversi dello stesso originator. Come ben noto, questa diversità dipende dal fatto che nel farmaco biotecnologico il prodotto (a suo tempo coperto da brevetto) non è la molecola che si ottiene dalla cellula, ma il processo di produzione che genera quella molecola. Ripetendo il processo produttivo due, tre, dieci, mille volte, il risultato non sarà mai identico pur trattandosi dello stesso farmaco.
Eppure, nella sentenza il Consiglio di Stato afferma che le differenze nel modello di glicosilazione sono sufficienti per escludere l’identità del principio attivo, ancorché il farmaco sia stato autorizzato come biosimilare (perché avente lo stesso profilo di sicurezza ed efficacia terapeutica. Si badi: lo “stesso profilo”, e non solo un “profilo equivalente”).
Avremmo cioè un tertium genus, che va ad aggiungersi a biosimilare, e ad originator diverso ma equivalente: lo strano caso del biosimilare avente principio attivo diverso.
La conclusione non convince: se il principio attivo è diverso, EMA non avrebbe potuto e dovuto autorizzare il farmaco con la procedura semplificata richiesta per i biosimilari. Avrebbe invece dovuto richiedere i risultati delle sperimentazioni di fase I, di fase II e clinica ed autorizzarlo come originator (diverso) ancorché terapeuticamente equivalente.
L’errore, metodologico, sta a mio avviso nell’aver enfatizzato le differenze rimarcate da AIFA tra epoetina zeta ed epoetina alfa (originator): quelle differenze sono proprie di ogni biosimilare ed anzi di ogni farmaco biologico.
Anche il secondo position paper di AIFA, pure utilizzato nella motivazione della sentenza, vengono messe in rilievo le differenze. Però non si può sorvolare sul fatto che: a) tali differenze sono definite come “minori”, nel senso che non sono tali da spezzare il rapporto di equivalenza (forte) del biosimilare per degradarlo a rapporto di equivalenza (debole) tra due principi attivi diversi; b) sono dovute al peculiare metodo di produzione dei farmaci biologici.












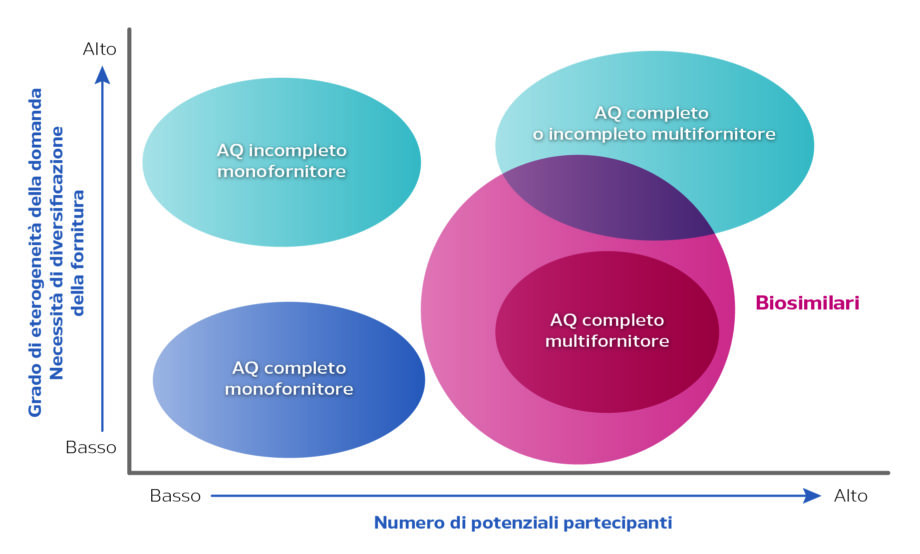
 Il ricorso all’accordo quadro è diventato più frequente da quando è stato reso obbligatorio per i farmaci biosimilari, con la Legge di Bilancio 2017, e fonte di confusione. Ad esempio il “tam tam” mediatico che si è innescato recentemente sugli acquisti dei biosimilari di adalimumab, e il susseguirsi sulla stampa di notizie in merito ai fortissimi sconti ottenuti da alcune Regioni, come ad esempio il Piemonte o la Toscana (che sono state le prime ad attivarsi) ne è un esempio. In realtà, questi acquisti così vantaggiosi non sono stati il frutto di un accordo quadro, perché, al momento della gara, non erano presenti sul mercato almeno 4 prodotti a base del principio attivo adalimumab, dunque non era obbligatorio per legge impiegare questa tipologia di procedura. In questi casi perciò le amministrazioni aggiudicatrici hanno indetto in realtà una gara senza accordo quadro e hanno acquisito non i primi 3 farmaci con la migliore offerta economica , come accade con accordo quadro, ma un solo farmaco, quello che ha fatto l’offerta più favorevole e mediamente stipulando un contratto a breve termine. In questo modo, quando diventano disponibili sul mercato altri biosimilari dello stesso principio attivo e si supera il numero di 3, è necessario, secondo la normativa vigente, prevedere una nuova gara, questa volta sì con un accordo quadro garantendo la disponibilità di 3 biosimilari diversi. Questo può avere importanti ricadute anche a livello clinico/organizzativo, perché i medici possono trovarsi nella condizione di dover sostituire le terapie appena prescritte ai pazienti a causa dei risultati della nuova gara: lo switch da un farmaco biosimilare ad un altro, anche se credo non costituisca un problema in termini di efficacia e sicurezza della terapia può però creare problemi perché i clinici debbono, ad ogni cambio di farmaco, informare il paziente, e se è presente un device per la somministrazione (come ad esempio una diversa penna) fornire le informazioni necessarie al suo corretto utilizzo, con un importante consumo di tempo. Tutto ciò ovviamente non favorisce l’uso del biosimilare. Non dimentichiamo che il reale risparmio di tale operazione non si realizza con l’acquisto, ma con la realizzazione degli switch.
Il ricorso all’accordo quadro è diventato più frequente da quando è stato reso obbligatorio per i farmaci biosimilari, con la Legge di Bilancio 2017, e fonte di confusione. Ad esempio il “tam tam” mediatico che si è innescato recentemente sugli acquisti dei biosimilari di adalimumab, e il susseguirsi sulla stampa di notizie in merito ai fortissimi sconti ottenuti da alcune Regioni, come ad esempio il Piemonte o la Toscana (che sono state le prime ad attivarsi) ne è un esempio. In realtà, questi acquisti così vantaggiosi non sono stati il frutto di un accordo quadro, perché, al momento della gara, non erano presenti sul mercato almeno 4 prodotti a base del principio attivo adalimumab, dunque non era obbligatorio per legge impiegare questa tipologia di procedura. In questi casi perciò le amministrazioni aggiudicatrici hanno indetto in realtà una gara senza accordo quadro e hanno acquisito non i primi 3 farmaci con la migliore offerta economica , come accade con accordo quadro, ma un solo farmaco, quello che ha fatto l’offerta più favorevole e mediamente stipulando un contratto a breve termine. In questo modo, quando diventano disponibili sul mercato altri biosimilari dello stesso principio attivo e si supera il numero di 3, è necessario, secondo la normativa vigente, prevedere una nuova gara, questa volta sì con un accordo quadro garantendo la disponibilità di 3 biosimilari diversi. Questo può avere importanti ricadute anche a livello clinico/organizzativo, perché i medici possono trovarsi nella condizione di dover sostituire le terapie appena prescritte ai pazienti a causa dei risultati della nuova gara: lo switch da un farmaco biosimilare ad un altro, anche se credo non costituisca un problema in termini di efficacia e sicurezza della terapia può però creare problemi perché i clinici debbono, ad ogni cambio di farmaco, informare il paziente, e se è presente un device per la somministrazione (come ad esempio una diversa penna) fornire le informazioni necessarie al suo corretto utilizzo, con un importante consumo di tempo. Tutto ciò ovviamente non favorisce l’uso del biosimilare. Non dimentichiamo che il reale risparmio di tale operazione non si realizza con l’acquisto, ma con la realizzazione degli switch. In Regione Sicilia abbiamo attivato alcune convenzioni tramite CONSIP ma non abbiamo mai effettuato gare aggregate per più Regioni. Le gare per l’acquisto di farmaci sono state effettuate sempre a livello regionale, tramite la Centrale Unica di Committenza (CUC).
In Regione Sicilia abbiamo attivato alcune convenzioni tramite CONSIP ma non abbiamo mai effettuato gare aggregate per più Regioni. Le gare per l’acquisto di farmaci sono state effettuate sempre a livello regionale, tramite la Centrale Unica di Committenza (CUC).