Intervista al Professor Giancarlo la Marca, Responsabile del Laboratorio di Screening Neonatale, Biochimica e Farmacologia, Ospedale Meyer
Come viene realizzato lo screening neonatale in Italia?
Oggi lo screening neonatale è considerato non un semplice test di medicina di laboratorio ma un programma complesso, multidisciplinare, che inizia prima della nascita del bambino, con l’informazione data alle famiglie e alle mamme durante il percorso nascita e la formazione del personale che deve raccogliere e maneggiare correttamente questo prelievo speciale. Il test di screening neonatale si effettua su alcune goccioline di sangue che, per obbligo di legge, in Italia vengono prelevate dal tallone del neonato tra le 48 e le 72 ore di vita e depositate su uno speciale supporto di carta. Come tutti i test di screening, anche quello neonatale non è diagnostico di per sé ma consente di selezionare una serie di neonati apparentemente sani che potrebbero sviluppare patologie rare conosciute come errori congeniti del metabolismo. In caso di test positivo, si procede ad un accertamento aggiuntivo di secondo livello per la conferma dello screening positivo, conferma che può essere biochimica o genetica. Quando si identifica definitivamente la malattia, il bambino viene preso in carico dal Servizio Sanitario Nazionale e dai centri clinici di riferimento per le malattie metaboliche.
Nel nostro Paese il panel di screening esteso include anche patologie la cui terapia è ancora in studio
Anche se l’evoluzione legislativa del sistema di screening italiano è stata estremamente significativa specialmente nell’ultimo periodo, la prima regolamentazione normativa risale al 1992, con la Legge 104 sulle disabilità, che rendeva obbligatorio lo screening obbligatorio per fibrosi cistica, ipotiroidismo congenito e fenilchetonuria. Dall’inizio del 2000, su un modello nato alla fine degli anni ’90 negli Stati Uniti, il numero di patologie si è esteso progressivamente in molti paesi industrializzati. La forte evoluzione che c’è stata in Italia è dovuta anche all’interesse che la politica ha manifestato per le malattie rare, a partire dalla Legge di stabilità del 2013, che ha dato il via ad una serie di iter normativi culminati con un finanziamento ad hoc e l’emanazione della Legge 167 del 2016 che oggi regolamenta lo screening in Italia, unica nazione in Europa, che offre obbligatoriamente uno screening per 40 malattie metaboliche ereditarie.
La copertura nazionale purtroppo non è ancora del 100%, e sicuramente questo è uno degli elementi da migliorare. La Legge 167 aveva l’obiettivo di evitare disuguaglianze all’interno del territorio nazionale: nonostante ciò, alcune Regioni sono partite con un certo ritardo e in questo momento dobbiamo ancora registrare la situazione della Calabria che non effettua questo servizio di medicina preventiva per i suoi circa 17.000 neonati l’anno. Come comunità scientifica, siamo al lavoro perché lo screening neonatale esteso possa partire anche in questa Regione dal prossimo anno.
Come dicevo prima, l’Italia è all’avanguardia. Siamo l’unica nazione europea ad aver normato lo screening neonatale come programma di prevenzione includendo anche la presa in carico del bambino da parte del SSN, quindi anche l’approccio terapeutico, che costituisce un elemento fondamentale del processo. Effettuare un’identificazione precoce della malattia non serve a nulla se poi il bambino non viene adeguatamente seguito e trattato precocemente per fare in modo che si modifichi la storia naturale della malattia e la vita del bambino possa veramente cambiare rispetto a prima, quando la diagnosi veniva posta solo dopo che il bambino cominciava a manifestare i sintomi della malattia e a stare male. Il sistema diventa efficiente se si riesce a porre la diagnosi quando il bambino sta bene, iniziando la terapia prima dell’insorgere dei sintomi.
Il sistema di screening neonatale genera risparmi a lungo termine per il SSN e migliora la qualità della vita
Inoltre, uno degli aspetti più importanti della legge, e un ulteriore passo avanti rispetto ad altri modelli internazionali è che, mentre tra i criteri universalmente riconosciuti perché una malattia venga inserita in un pannello di screening c’è quello che debba esistere già una terapia disponibile e accessibile, la Legge 167 prevede tra i suoi articoli di poter inserire nel pannello di screening anche quelle patologie per cui la terapia dietetica o farmacologica sia ancora in studio, come ad esempio un trial clinico in stato avanzato. In questo senso, l’identificazione precoce permette ai neonati di accedere a queste sperimentazioni in modo molto efficiente e rappresenta un grosso vantaggio per le terapie più innovative.
Lo screening neonatale esteso, che intende favorire l’accesso ai pazienti a terapie avanzate e innovative e incrementare il numero dei pazienti in trattamento, può risultare sostenibile anche dal punto di vista economico per il SSN?
Il quesito si pone sicuramente, e la risposta è un po’ complessa. Il punto di partenza, che riguarda tutti i farmaci orfani e per le malattie rare, è il costo iniziale spesso molto consistente di questo tipo di terapie. Tanto consistente che è impossibile pensare che possa essere a carico della famiglia del paziente, specialmente quando richiede una somministrazione ripetuta nel tempo. Il costo molto elevato rappresenta un elemento di criticità molto forte che può costituire uno dei motivi principali per i sistemi economici, amministrativi o anche per i Comitati etici, principalmente fuori dall’Italia, per non allargare il proprio pannello di screening.
Il modello italiano è riconosciuto all’avanguardia anche in termini di accessibilità ai farmaci
In realtà l’elemento costo del sistema screening deve essere visualizzato e analizzato correttamente e nel suo complesso prendendo in considerazione tutto il contesto: anche in assenza di programmi di screening neonatale precoce, i bambini affetti da queste patologie nascono lo stesso, però vengono indirizzati alla terapia in maniera tardiva. Il ricorso al farmaco costoso c’è comunque, anche se viene iniziato più tardi. Con lo screening neonatale, è possibile identificare quel bambino con quella stessa malattia in una fase migliore e più precoce, in cui la patologia può non aver ancora causato danni consistenti. È vero che, iniziando prima la terapia, anche i costi aumentano, ma si tratta di un circolo virtuoso perché il trattamento terapeutico intrapreso precocemente permette un miglioramento della qualità di vita, con guadagni in termini di salute ed economici per il bambino malato e per tutta la sua famiglia. Laddove sono stati effettuati studi economici sulla singola malattia, ad esempio sui deficit di ossidazione degli acidi grassi, come il deficit di MCAD, è stato dimostrato che la prevenzione fa risparmiare moltissimo al sistema sanitario nazionale. Questa considerazione è stata alla base delle scelte che hanno permesso all’Italia di accelerare il passo su questo tema: quando c’è stata la modifica della legge, io ho avuto la fortuna di essere tra gli esperti interpellati e posso documentare che questo elemento ha assunto un ruolo chiave. Alla domanda su quanto costa il sistema di screening neonatale, avanzata dalla politica e dalla pubblica amministrazione, la risposta migliore è stata la dimostrazione dei risparmi che il sistema screening era in grado di generare. Il risparmio purtroppo non è mai immediato, perché il paziente deve essere seguito nel tempo ma se il sistema funziona con efficienza i risparmi si verificano. In realtà ad oggi, ciò che costituisce un possibile aumento di costi per il SSN non sono i veri malati ma i falsi positivi (soggetti positivi al test di screening ma sani) che vengono indirizzati ai controlli di secondo livello ma non risultano affetti dalla patologia: su questo punto il processo può essere sicuramente migliorato.
Per quanto riguarda le terapie per le patologie individuate con lo screening neonatale, com’è la situazione in Italia? Sono disponibili, e in che tempi?
Uno dei criteri con cui sono state scelte le 40 patologie per lo screening neonatale esteso è che per ognuna ci deve essere un trattamento terapeutico, sia esso dietetico o farmacologico. Quindi, le terapie oggi esistono. L’effettiva disponibilità sul territorio e le relative tempistiche di erogazione dipendono invece dall’organizzazione delle singole Regioni: in alcuni casi, il servizio farmaceutico regionale può contare su una scorta dei farmaci necessari mentre in altri casi, per una serie di motivi solitamente economici o di organizzazione regionale, non sono previste scorte e si devono attivare iter appositi, a seguito della diagnosi, con il conseguente slittamento della terapia. Anche su questo aspetto, la comunità scientifica e clinica può attivarsi per fare meglio.
L’effettiva disponibilità sul territorio è ancora disomogenea e su questo la comunità scientifica è al lavoro
In conclusione, vorrei sottolineare che, anche in termini di accessibilità dei farmaci, tutto il mondo ci invidia questo modello di gestione efficiente dello screening neonatale e la possibilità aggiuntiva che una patologia sia inseribile in un pannello di screening neonatale anche se la terapia dietetica o farmacologica è ancora in via di sviluppo. In Italia noi siamo stati e siamo ancora oggi pionieri di molti trattamenti terapeutici innovativi, si pensi ai successi sulla terapia genica ottenuti dagli Istituti Telethon di Napoli e Milano. Questi traguardi dei colleghi ricercatori sono per noi fondamentali perché ci hanno permesso e ci permetteranno di essere all’avanguardia anche per la parte diagnostica in quanto le nuove terapie sono un continuo stimolo anche alla nostra ricerca e questa combinazione, che è il modo più efficiente di fare buona salute, ancora oggi ci consente di essere un passo avanti alle altre nazioni europee.
Intervista a Francesco Macchia, Editore Osservatorio Terapie Avanzate (OTA)
Che cosa si intende con il termine “terapie avanzate”?
Per parlare correttamente di “terapie avanzate” si dovrebbe utilizzare il termine inglese “Advanced therapy medicinal products (ATMP)”, definito dal Regolamento CE n. 1394 del 2007 che ha istituito il Committee for Advanced Therapies (CAT) dell’EMA, ovvero il Comitato che decide a livello europeo l’approvazione delle terapie avanzate. Con il termine ATMP si intendono quelle terapie o farmaci innovativi basati sulla terapia genica, la terapia cellulare e l’ingegneria tissutale. Si differenziano pertanto sia dai farmaci tradizionali, basati su molecole prodotte per sintesi chimica, sia dai farmaci biologici quali ad esempio gli anticorpi monoclonali. In questo momento rappresentano delle terapie totalmente “breakthrough” perché saranno destinate, nei prossimi anni, a stravolgere completamente il concetto di medicina e di terapia.
La classificazione delle terapie avanzate è un tema piuttosto complesso, e non esiste una classificazione standardizzata universalmente condivisa. L’Osservatorio Terapie Avanzate (OTA) con il suo Comitato Scientifico ha elaborato una classificazione dall’intento anche divulgativo, suddividendo le terapie avanzate in: prodotti a base di DNA e cellule, ma anche di piccole molecole di RNA o di tecnologie per la manipolazione del genoma o la creazione di organoidi. Rientrano tra i prodotti di terapia genica quelli che hanno l’obiettivo di trattare malattie causate da geni difettosi: il farmaco è costituito direttamente dal “gene terapeutico” (DNA) o da un sistema in grado di correggere le mutazioni, un esempio è l’editing genomico con CRISPR-cas9. Fanno parte invece della terapia cellulare i prodotti a base di cellule vive per ottenere un effetto terapeutico, diagnostico o preventivo, come ad esempio le cellule staminali adulte. La terza categoria comprende il mondo dell’ingegneria tissutale, in prospettiva molto sfidante: si tratta di cellule, tessuti e organi che possono essere ricostruiti in vitro e trapiantati all’interno dell’organismo. E siccome la scienza è in continua evoluzione, è già stata introdotta anche una quarta categoria che comprende approcci che combinano insieme le terapie avanzate precedentemente definite.
Quali sono le peculiarità delle terapie avanzate rispetto ai farmaci tradizionali?
Per chiarire la portata innovativa delle terapie avanzate rispetto alle terapie tradizionali bisogna considerare fondamentalmente due tematiche: innanzitutto, per la prima volta, si passa da un approccio “terapeutico” ad un approccio “curativo”. Già con la terapia innovativa per l’HPV c’è stato un primo approccio curativo che ha consentito di parlare di “eradicazione” della malattia in tempi brevi, ma per le terapie avanzate l’approccio curativo costituisce una caratteristica intrinseca e imprescindibile. Non si tratta più di una terapia ma di una vera e propria cura che eradica alla base l’espressione sintomatica della malattia. Il secondo aspetto, altrettanto straordinario e rivoluzionario, è che le terapie avanzate sono “one-shot” o, laddove debbano essere ripetute 2-3 volte, il trattamento rimane comunque molto limitato nel tempo mentre i suoi effetti si prolungano nel lungo periodo.
Quali difficoltà comporta l’accesso a tali terapie? Quali soluzioni si stanno cercando?
L’approccio curativo e la somministrazione one-shot cambiano completamente l’ottica di gestione della malattia rispetto alle terapie tradizionali e anche rispetto ad alcuni farmaci innovativi, come ad es. gli anticorpi monoclonali. È chiaro che questo cambiamento di paradigma porta con sé una serie di complessità e di difficoltà. Come valorizzare un costo e un prezzo di terapia su processi totalmente innovativi? Questa è una sfida per tutto il sistema: in Europa su questo tema si sta lavorando alacremente e, ad esempio, in Italia è recente la notizia della rimborsabilità delle CAR-T tramite “payment at results”. Le difficoltà in merito all’accesso alle terapie avanzate sono numerose e strettamente correlate alle loro caratteristiche intrinseche. Innanzitutto siamo dinanzi ad un livello di complessità del meccanismo d’azione enorme rispetto alle terapie tradizionali: in questo senso, se già il funzionamento di un farmaco biologico è estremamente complicato rispetto ad un antibiotico, la terapia genica rappresenta un salto tecnologico straordinario.
L’aspetto della manifattura è anch’esso estremamente complicato in quanto spesso coinvolge materiale biologico del paziente che deve essere estratto, messo in coltura, a volte manipolato geneticamente e ogni step deve seguire le procedure GMP. La somministrazione delle terapie avanzate è un altro elemento di complessità intrinseca poiché sono necessariamente pochi e altamente specializzati i centri in grado di trattare i pazienti con questa tipologia di terapia. E poi ci sono i costi per la somministrazione di questi prodotti che, come noto, sono altissimi.
In questo panorama già così complesso si introduce un ulteriore elemento di difficoltà, che deriva anch’esso dalle caratteristiche intrinseche delle terapie avanzate ed è rappresentato dalla “relativa” scarsità delle prove di efficacia. Sono due gli elementi che vi concorrono: innanzitutto, poiché si tratta di farmaci innovativi, in genere EMA riconosce loro delle modalità di Early Access Programme per l’autorizzazione al commercio. Questo percorso, logico e razionale dal punto di vista EMA, complica però la situazione al momento della definizione di prezzo e rimborso a livello delle singole Agenzie Regolatorie degli Stati. Inoltre, pur trattandosi di terapie curative e dunque potenzialmente risolutive di una patologia, i dati di efficacia a lungo termine non sono ancora disponibili. E la maggior parte riguarda trattamenti dedicati a malattie rare, che per definizione possono contare su dati relativi a popolazioni limitate.
L’approccio curativo e la somministrazione one-shot rendono “rivoluzionarie” le terapie avanzate
È chiaro dunque che le difficoltà di accesso sono enormi, soprattutto perché, nonostante l’intenso lavoro sia di EMA sia delle Agenzie regolatorie nazionali, il sistema non è ancora pronto: basti pensare alla valutazione tramite il sistema GRADE, che è costruito sulle terapie tradizionali, in cui il livello di evidenza è la base.
Come si può garantire un efficace early access per i pazienti, sostenendo allo stesso tempo l’importante impatto economico di queste terapie?
Da questo punto di vista oggi abbiamo un modello su cui ragionare e che può darci già qualche indicazione utile, ovvero l’accordo tra Novartis e AIFA su Kymriah, sulle CAR-T per il linfoma diffuso e la leucemia linfoblastica acuta. A mio parere, questo accordo può rappresentare un modello di governance delle terapie avanzate. Il punto chiave riguarda l’applicazione di una formula di pay-for-performance denominata “payment at results” che prevede il rimborso in base ai risultati ottenuti nella pratica clinica e non in base a quanto dimostrato nei trial clinici. Questo approccio può essere applicato efficacemente per i farmaci ad alto costo e soprattutto in presenza di evidenze non così solide da supportare un accordo di pagamento tradizionale. In questo senso l’accordo su Kymriah è innovativo in Europa, anche se in Italia questa modalità di pagamento era già stata introdotta da Nello Martini come primo Direttore generale di AIFA.
Le diverse formule di pay-for-performance possono essere utilizzate solo se combinate con un sistema di Real World Evidence basato su registri di monitoraggio affidabili e per paziente; forse proprio per questo il payment at results non è molto comune a livello europeo, mentre può essere applicato nel nostro Paese grazie al sistema di registri attivo e funzionante con un approccio unitario grazie al Servizio Sanitario Nazionale.
Un altro elemento fondamentale riguarda le modalità di pagamento, che dovranno prevedere una rateizzazione anche tramite modelli assicurativi o di finanziamento bancario ancora da definire. Se pensiamo che per alcune terapie avanzate allo studio si parla di costi da 1 milione di euro a paziente, è evidente che lo Stato non potrà sostenere da solo e nell’immediato un tale impegno.
Da ultimo bisogna sottolineare come probabilmente, nei prossimi anni, i fondi per i farmaci innovativi (oncologici e non) potrebbero non essere sufficienti a coprire le spese relative alle terapie avanzate, pertanto si dovrà pensare a fondi ad hoc che consentano innanzitutto di garantire un accesso rapido a queste terapie e soprattutto di renderle disponibili senza distinzioni su base regionale.
Come si pongono le terapie avanzate nel sistema di early access in Italia?
Nel nostro Paese le normative che regolano l’early access sono sicuramente all’avanguardia, anche nel contesto internazionale ed europeo, e sono in grado di garantire un rapido accesso ai pazienti; devono però essere adattate e “tarate” sulle peculiarità delle terapie avanzate. Un primo elemento che andrebbe modificato è l’introduzione di un prezzo concordato (al momento i farmaci che beneficiano dell’early access in Italia vengono immessi in commercio con il prezzo stabilito dall’azienda). Quindi si dovrebbero considerare anche elementi più tecnici, come la possibilità di fare riferimento ad endpoints surrogati (per sopperire, come dicevamo, alla mancanza di dati di efficacia a lungo termine) e di sviluppare ulteriori modalità di confronto indiretto (per far fronte alla difficoltà di condurre sperimentazioni versus placebo).
Quali sfide ci pongono le future terapie?
In termini prospettici, la grande sfida riguarda la sostenibilità di sistema. Nel tempo, se è vero che i costi di ricerca e sviluppo per le terapie avanzate andranno a ridursi (grazie alla standardizzazione di alcuni processi), è anche vero che tali terapie verranno utilizzate sempre meno su malattie rare e sempre più per malattie ad ampia diffusione, con un nuovo incremento della spesa. Secondo me, la capacità di cogliere questa sfida non riguarderà tanto le modalità di contrattazione dei prezzi quanto piuttosto la capacità dell’Europa e dell’Italia di sfruttare al meglio le grandi opportunità di crescita tecnologica, scientifica e manufatturiera del settore, per diventare un polo non solo di ricerca, ma anche di sviluppo e produzione di terapie avanzate.
In quest’ottica, ad esempio, è attivo a livello europeo RESTORE, che vede in Italia Fondazione Telethon come partner e Osservatorio Terapie Avanzate come supporter. RESTORE è un’iniziativa di ricerca portata avanti da dieci realtà europee leader nel settore delle terapie avanzate che propone da un lato di trasformare la promessa delle terapie avanzate in un’opportunità concreta per i pazienti e dall’altro punta a far sì che l’Europa mantenga un ruolo di primo piano nel settore, restando competitiva sia nel campo della ricerca che in quello dello sviluppo. Un importante obiettivo è di costruire un ecosistema che sia sostenibile nel suo complesso e non solo in termini di equilibrio di bilancio a livello di fondi.
Intervista al Professor Luca Pani, Università di Modena e Reggio Emilia, University of Miami
Quali sono le peculiarità delle terapie avanzate che richiedono nuovi modelli di pricing e rimborsabilità?
La definizione di terapie avanzate dal punto di vista regolatorio è abbastanza chiara ma le innovazioni tecnologiche degli ultimi 5 anni rappresentano delle sfide per coloro che devono approvare e soprattutto dare il giusto valore a tanta presunta innovazione. Il punto importante da sottolineare subito è infatti che l’innovazione resterà teorica sino a quando non avremmo abbastanza dati per valutare l’impatto di terapie eradicanti e che richiedono una singola somministrazione, sulla progressione e la modifica di malattie altrimenti fatali. Tanto per concentrarci, da subito, sul tema più importante: quello di prodotti teoricamente capaci di fare la differenza tra una vita normale o morte sicura.
Il modello basato su 3 prezzi consente di modulare la rimborsabilità anche in base al livello di evidenza di efficacia disponibile
L’anno scorso lei ha pubblicato un nuovo modello di pricing, elaborato con un gruppo di economisti: in che cosa consiste?
Nelle condizioni che ho appena descritto ci si trova a negoziare, sia da punto di vista del pagatore pubblico o privato (i.e. assicurazioni sanitarie) che dell’industria, in condizioni di estrema incertezza e sotto altissime pressioni psicologiche e sociali che troppo spesso diventano politiche. In questi casi la strategia di attendere o, peggio, rifiutarsi di discutere un prodotto farmaceutico con una efficacia potenziale superiore alle terapie standard non è eticamente accettabile ma non lo è neppure ammettere delle rimborsabilità sino a milioni di euro per paziente senza conoscere per quanto a lungo la nuova terapia avrà effetto o anche ignorare per quanti pazienti sarà necessaria. Abbiamo quindi proposto un modello basato su tre prezzi: il primo molto basso quando le evidenze di efficacia non sono ancora certe; un secondo variabile, ma più alto del primo, deciso in base ai risultati della prima fase e un terzo prezzo che torna a livelli più bassi e predeterminati pur in presenza di una protezione brevettuale attiva per consentire a nuovi prodotti innovativi, magari della stessa azienda, di entrare sul mercato con lo stesso tipo di schema e dunque arrivare prima ai pazienti.
Quali sono i principali vantaggi e le criticità, rispetto ad altri modelli come ad esempio il pay-for-performance?
I vantaggi sono quelli che ho appena illustrato: far accedere a terapie realmente miracolose in tempi brevissimi, da 3 a 5 anni prima, di quanto accade adesso. Un altro vantaggio emerge quando si tratta di farmaci che curano popolazioni di pazienti dalla epidemiologia imprecisa, in cui non sappiamo esattamente quanti siano, il modello in questo caso consente di ridurre l’impatto economico di questa incertezza. La criticità maggiore, che di fatto lo rende prevalentemente teorico, è che nessun pagatore – pubblico o privato – accetterebbe di aumentare il rimborso di un prodotto che, per anni, ha pagato anche cinque volte di meno. A meno che non si possano applicare dei correttivi su cui infatti stiamo lavorando.
A suo parere, questo modello è applicabile alla realtà italiana, e in che termini?
Sylvain Chassang e Erik Snowberg, i due economisti con cui abbiamo elaborato queste idee e con cui ancora collaboriamo, furono attratti proprio dalle strategie negoziali dell’AIFA tra la fine del 2011 e il 2016. Questo modello nasce quindi dalla realtà italiana ovvero dall’uso “illuminato” che, in quel periodo, facemmo di centinaia di contratti negoziali vincolati a registri certificati e web based (certo non cartacei) e al payment by result cancellando – di fatto – schemi assolutamente obsoleti di risk e cost sharing che, per questo tipo di prodotti e per quelli che arriveranno, sono completamente inadeguati.
L’obiettivo è garantire l’accesso ai pazienti in tempi molto brevi e compensare le incertezze delle terapie innovative
Questo modello potrà favorire l’early access delle terapie avanzate ai pazienti?
È stato pensato esattamente per questo motivo ma anche per compensare le incertezze che sono insite nelle cosiddette innovazioni terapeutiche, molte delle quali potrebbero non rivelarsi tali. Tuttavia, e nonostante questa premessa, bisogna tenere presente che nei prossimi 8-10 anni ci saranno da valutare e negoziare circa 1.000 terapie avanzate e che, se pure ne fallissero il 90%, qualcosa che nessuno si augura per le speranze dei malati, i nostri sistemi di rimborsabilità attuali non saranno in grado di renderle disponibili a chi ne avrà bisogno. Abbiamo l’obbligo morale di trovare dei modelli alternativi che consentano l’accesso e che siano non solo scientificamente ed economicamente solidi e sostenibili ma anche realistici e logici da attuare. Stiamo lavorando per fare in modo che questo sia possibile.
Intervista al Professor Mario Melazzini, Direttore Scientifico Centrale degli Istituti Clinici Maugeri, Pavia
Alcuni studi evidenziano come i tempi di accesso ai farmaci dopo l’approvazione in EMA possono essere piuttosto lunghi: le norme italiane sull’early access riescono ad incidere positivamente sulla tempestività di accesso per i pazienti?
Dentro ogni norma si muove il lavoro delle persone. La normativa italiana ha dimostrato nel corso del tempo di riuscire a raccogliere la sfida della velocità che, riferita alla vita delle persone e al loro legittimo bisogno di salute, si è puntualmente tradotta in modalità che permettano di accedere alle terapie nel più breve tempo possibile. E quando arrivano nuove terapie bisogna farsi trovare pronti, con attività che cominciano ben prima della negoziazione del prezzo. Garantire ai pazienti un accesso rapido ai nuovi farmaci nel rispetto delle evidenze scientifiche che assicurino qualità, efficacia e sicurezza deve essere sempre la direttrice delle attività regolatorie italiane. E finora c’è stata, attivando strumenti diversi.
Pensiamo al cosiddetto fondo del 5%: solo nel 2017 il nostro Paese aveva impegnato quasi venti milioni di euro. Una cifra enorme ma indispensabile per rispondere a bisogni di salute sempre diversi e crescenti. Nel nostro Paese esiste la possibilità di ricorrere al cosiddetto “uso compassionevole” che consente di utilizzare un medicinale sottoposto a sperimentazione clinica, al di fuori della sperimentazione stessa, in pazienti affetti da malattie gravi o rare o che si trovino in pericolo di vita laddove, per esempio, non vi siano ulteriori valide alternative terapeutiche. Esiste anche la possibilità di accedere a un farmaco tramite la Legge 648/1996 che consente, quando non esista un’alternativa terapeutica valida, di erogare un farmaco autorizzato in altri Stati, ma non in Italia, di erogare medicinali non ancora autorizzati, ma in corso di sperimentazione clinica e medicinali da impiegare per una indicazione terapeutica diversa da quella autorizzata. Insomma nel nostro Paese il legislatore ha dimostrato quasi sempre coraggio.
Quali sono le principali criticità degli attuali canali dell’early access italiani per la gestione delle terapie innovative o avanzate?
Sono tre le principali criticità legate agli accessi precoci, indipendentemente dai canali normativi o dalle fasi storiche: il valore relativo dei dati preliminari, le evidenze negative che possono palesarsi in una fase successiva dello sviluppo e, da un punto di vista più etico, l’applicazione forzata del concetto di unmet medical need e di uso compassionevole. La storia ci ha dimostrato purtroppo come anche le situazioni più drammatiche possano essere strumentalizzate o comunque utilizzate in modo distorto e dannoso.
Nella sua esperienza, quali strumenti sono maggiormente utilizzati ed efficaci per garantire l’accessibilità in tempi rapidi dei farmaci ai pazienti e quali potrebbero essere migliorati? Potrebbe essere utile una riforma legislativa di alcuni strumenti, e in che termini (es. Legge 648 o classe Cnn)?
L’Italia ha sempre mostrato una sensibilità unica nel panorama internazionale, ma credo che in termini di accessibilità e strumenti di valutazione del tempo sarebbe auspicabile un confronto serio per valutare se e quali siano le realtà nazionali che riescono a razionalizzare al meglio le procedure di autorizzazione. Prima di un confronto europeo però credo sarebbe opportuno fare un’analisi delle tante, troppe differenze regionali, che non possiamo fare finta di non vedere. Persistono delle diseguaglianze, infatti, che andrebbero affrontate con rigore e onestà. La segmentazione del potere di intervento, statale, regionale, locale non può reggere senza una cabina di regia politica e amministrativa in grado di fare valutazioni nazionali efficienti. Non basta fare la propria parte, non è sufficiente.
Sul piano dei costi, quali potrebbero essere le strategie da mettere in atto per garantire la sostenibilità di terapie innovative, come le CAR-T o la terapia genica, nell’ambito del Servizio Sanitario Nazionale?
Sicuramente non da oggi, ma oggi più che mai i dati sono il cuore di ogni passo, sia questo di programmazione sia di valutazione. Mentre sul piano privato collezionare informazioni è normale perché evita duplicazioni e massimizza informazioni anche di secondo livello, il mio timore è che i policy maker non abbiano ancora colto l’importanza strategica delle banche dati. Sottolineo questo passaggio perché dalle informazioni pubbliche mi pare che attualmente non esista ancora un database europeo per le terapie CAR-T, il che significa che, inconsapevolmente o meno, più persone potrebbero stare lavorando a progetti simili, perdendo così la possibilità di ottimizzare risorse strumentali, finanziarie e umane. La sostenibilità passa anche attraverso un’allocazione efficace ed efficiente di risorse, da un punto di vista industriale ma soprattutto statale.
La normativa italiana ha raccolto la sfida della velocità ma le differenze regionali sono ancora troppo marcate
Ci sono altre esperienze, a livello europeo e internazionale, che potrebbero offrire degli spunti per migliorare accessibilità e sostenibilità in Italia?
L’Italia si è sempre contraddistinta in questo campo, siamo stati esempio e abbiamo contributo a generare condizioni di grande competitività a vantaggio dei nostri cittadini, dei pazienti. Il confronto aperto è sempre una ricchezza, ma il modello italiano deriva da una Carta Costituzionale che dobbiamo tenerci stretta: l’obbligo di garantire la salute anche attraverso la dispensazione dei farmaci è la bussola. Poi di esempi possiamo trarne anche altrove perché, come mi capita di dire spesso, c’è sempre la possibilità di migliorare, basta guardare le sfide e affrontarle con coraggio. C’è sempre un grande futuro ad aspettarci se solo sappiamo preparare al meglio il suo arrivo.
Negli ultimi anni il progresso in campo farmaceutico ha effettuato notevoli passi avanti, portando all’introduzione di farmaci sempre più innovativi e all’avvento dei farmaci biologici e della medicina personalizzata. La velocità con la quale vengono studiate e sviluppate le nuove alternative terapeutiche viene, però, frenata da procedure approvative lunghe e complesse che ritardano l’accesso dei pazienti ai nuovi trattamenti, talvolta salvavita.
Secondo un recente rapporto IQVIA pubblicato da EFPIA (European Federation of Pharmaceutical Industries and Associations), infatti, un nuovo farmaco è disponibile per i cittadini europei dopo una media di 426 giorni dall’approvazione in EMA (European Medicines Agency). Lo studio ha preso in esame 121 prodotti approvati con procedura centralizzata tra gennaio 2015 e dicembre 2017 e ha valutato il tasso di disponibilità (inteso come il numero di farmaci disponibili ai pazienti europei alla data del 19 dicembre 2018) e ritardo di accesso al mercato (inteso come il numero di giorni trascorsi dall’autorizzazione europea al termine delle procedure di autorizzazione nazionali) in 30 paesi europei.
In Europa il tempo medio di accesso al mercato di un nuovo farmaco è 426 giorni dall’approvazione in EMA
In termini di disponibilità dei farmaci l’Italia si colloca al quinto posto con il 79% dei 121 prodotti approvati in EMA disponibili alla data di cut-off dell’analisi. In testa alla classifica si trovano Regno Unito (88%), Germania (86%) e Austria (84%), mentre nelle ultime posizioni ci sono Macedonia (24%), Serbia (36%) e Lettonia (23%).
Per quanto riguarda i tempi di accesso al mercato l’Italia risulta meno virtuosa collocandosi al 14esimo posto, con una media di 402 giorni. Al primo posto si trova la Germania (119 giorni), seguita da Danimarca (146 giorni) e Svizzera (171 giorni), mentre in fondo alla classifica ci sono Serbia (925 giorni) e Lituania (726 giorni). La sottoanalisi, che ha preso in esame solo i farmaci oncologici, colloca l’Italia al 7° posto in termini di disponibilità con l’81% dei farmaci a disposizione e una durata media del processo di approvazione nazionale di 368 giorni (12° posto). Per quanto riguarda i farmaci orfani, invece, il tasso di disponibilità per l’Italia è del 76% (5° posto) mentre il tempo medio di approvazione è di 428 giorni (12° posto).
In Europa, a livello centralizzato e locale, sono numerose le iniziative intraprese negli ultimi anni per accelerare i tempi di accesso al mercato di farmaci che rispondono a importanti unmet needs, cercando di superare le incertezze in termini di efficacia e sicurezza derivanti dall’approvazione di molecole in fasi precoci di sviluppo.
Iter di approvazione di un nuovo farmaco
In Europa
Il Regolamento CE n. 726/2004, per l’istituzione di procedure comunitarie per l’autorizzazione e la sorveglianza dei medicinali per uso umano e veterinario, ha stabilito la necessità di rendere obbligatoria la procedura centralizzata di autorizzazione all’immissione in commercio (AIC), e la disponibilità immediata dopo l’approvazione in tutti gli stati membri, per tutti i medicinali:
ad alta tecnologia, in particolare quelli derivati da biotecnologie;
per terapia avanzata, quali la terapia genica, le terapie cellulari associate e la terapia somatica xenogenica;
orfani;
contenenti nuove sostanze attive aventi come indicazione terapeutica il trattamento della sindrome da immunodeficienza acquisita, del cancro, di disordini neurodegenerativi, del diabete, delle malattie autoimmuni e delle malattie virali.
È inoltre previsto l’accesso facoltativo alla procedura centralizzata quando il ricorso a una procedura unica porti netti vantaggi ai pazienti come nel caso di medicinali che rappresentano innovazioni terapeutiche o medicinali non innovativi che possono essere utili alla società o ai pazienti.
La procedura centralizzata ha inizio con la presentazione, da parte dell’azienda farmaceutica, della domanda di autorizzazione all’EMA a cui fa seguito, entro 210 giorni dal deposito del dossier, il rilascio del parere del CHMP (Committee for Medicinal Products for Human Use). In caso di parere positivo la decisione viene ratificata dalla Commissione Europea e pubblicata in Gazzetta Ufficiale dell’Unione Europea (GUUE) (Figura 1).
Figura 1. Iter approvativo di un nuovo farmaco dalla domanda di autorizzazione in EMA con procedura centralizzata alla disponibilità a livello locale italiano
In Italia
Mentre nella fase di approvazione europea l’EMA valuta l’efficacia, la sicurezza e la qualità del farmaco, a livello nazionale l’AIFA deve valutare il place in therapy della terapia nel contesto italiano, il rapporto di costo-efficacia e la sostenibilità per il SSN. Per ottenere la classificazione ai fini della rimborsabilità i farmaci autorizzati a livello europeo vengono pertanto sottoposti alla procedura negoziale con AIFA, che ha inizio con la presentazione del dossier di prezzo e rimborso (P&R).
Strumenti di Early Access
Legge 189/2012: prevede che un nuovo farmaco, entro 60 giorni dalla pubblicazione dell’AIC comunitaria in GUUE, venga classificato in classe Cnn e possa essere commercializzato prima della conclusione della procedura negoziale in AIFA. Criticità. La classificazione Cnn, nata con l’intento di rendere immediatamente disponibile un farmaco dopo l’approvazione in Europa, nei fatti spesso si è tradotta in un ostacolo all’accesso in quanto, non prevedendo il rimborso da parte del SSN, l’acquisto del farmaco è a totale carico del cittadino o delle singole strutture ospedaliere o ASL.
Legge 98/2013: stabilisce carattere prioritario per i farmaci orfani, quelli di eccezionale rilevanza terapeutica o sociale e a uso esclusivo ospedaliero e prevede che per questi farmaci l’azienda possa presentare il dossier P&R subito dopo la pubblicazione in GUUE, rinunciando all’inserimento in Cnn. Ha inoltre previsto che in questi casi l’iter negoziale debba terminare entro 100 giorni dalla presentazione della domanda.
Dopo la presentazione del dossier di P&R la Commissione Tecnico Scientifica (CTS) determina il rapporto di costo-efficacia ed esprime un parere, non vincolante, sulla classificazione del farmaco ai fini della rimborsabilità. In seguito a parere positivo della CTS, il Comitato Prezzo e Rimborso (CPR) stabilisce il valore economico del farmaco e imposta la contrattazione con l’azienda. Al termine della contrattazione, dopo aver raggiunto l’accordo su prezzo, classe di rimborsabilità ed eventuali condizioni contrattuali di accesso (registro di monitoraggio, piano terapeutico, MEA – Managed Entry Agreement), il Consiglio di Amministrazione (CdA) di AIFA ratifica la decisione e la Determina di prezzo e rimborso viene pubblicata in GU (Figura 1).
A questo punto, perché il farmaco sia accessibile ai pazienti è necessaria un’ulteriore valutazione a livello regionale. In accordo con la Legge 405/2001, che affida alle Regioni il mandato di attuare le iniziative volte al contenimento dei tetti di spesa, le realtà locali hanno istituito il Prontuario Terapeutico Regionale (PTR) e il Prontuario Terapeutico Ospedaliero Regionale (PTOR) nel quale sono inseriti i farmaci prescrivibili all’interno dei presidi ospedalieri regionali. La maggior parte delle Regioni ha adottato un PTOR vincolante, obbligando le strutture a prescrivere unicamente i farmaci presenti nel prontuario regionale, mentre altre Regioni non hanno adottato un PTOR e il farmaco è disponibile per la prescrizione dopo la pubblicazione della Determina AIFA (Figura 1).
Strumenti di Early Access
Legge 189/2012: stabilisce che i farmaci innovativi o a innovatività condizionata vengano inseriti immediatamente nei Prontuari Terapeutici Regionali.
Tempi di approvazione
Un’analisi sulle tempistiche di approvazione dei farmaci specifica sull’iter approvativo italiano è stata condotta da Lidonnici et al., che hanno quantificato i tempi impiegati per compiere i diversi step necessari affinché la Determina di P&R di un nuovo farmaco venga pubblicata in GU, dopo l’approvazione a livello europeo.
Dopo l’approvazione europea, la durata media dell’iter approvativo in Italia è di 258 giorni
Lo studio ha evidenziato che, dei 190 farmaci approvati in Europa tra gennaio 2015 e gennaio 2018, 85 hanno concluso l’iter approvativo in Italia con pubblicazione in GU della determina di P&R.
I tempi medi dall’approvazione CHMP e la pubblicazione in GUUE e la successiva pubblicazione in GU della classificazione in Cnn sono stati di 62 e 126 giorni, rispettivamente. La durata media dell’iter approvativo, dall’apertura della procedura in CTS alla pubblicazione della determina in GU, è stata invece di circa 258 giorni (73 giorni tra l’apertura della procedura e il parere della CTS, 94 giorni tra il parere della CTS e quello della CPR e 92 tra il parere della CPR e la pubblicazione in GU della determina AIFA) a cui si aggiungono 94 giorni tra l’applicazione della domanda e l’apertura della pratica in CTS, cioè il tempo necessario per la verifica delle informazioni fornite e l’istruzione della pratica.
Come riportano gli Autori, è interessante notare come il parere sul place in therapy del farmaco e sulla relativa rimborsabilità viene rilasciato da AIFA dopo circa 167 giorni dalla presentazione della domanda, tempi relativamente rapidi che potrebbero essere sfruttati per sviluppare un modello di early access in cui il farmaco viene messo a disposizione del SSN prima della contrattazione del prezzo.
Le sottoanalisi dello studio di Lidonnici et al., condotte su diverse tipologie di farmaci, hanno inoltre evidenziato tempistiche minori per la valutazione dei farmaci innovativi, rispetto ai non innovativi (196 vs 272 giorni) e tempi medi superiori per i farmaci orfani, rispetto ai non orfani (280 vs 251) e per i farmaci oncologici-oncoematologici, rispetto a quelli di altre aree terapeutiche (326 vs 234).
Un’analisi analoga, condotta da Prada et al., si è concentrata sui farmaci oncologici e ha quantificato il tempo trascorso tra la pubblicazione dell’AIC in GUUE e la data del primo acquisto a livello ospedaliero per i farmaci approvati in AIFA tra gennaio 2013 e dicembre 2015. Nel periodo in esame l’EMA ha approvato 34 nuovi farmaci oncologici, 14 dei quali con designazione di farmaco orfano. A luglio 2016, solo 17 erano disponibili sul mercato italiano e il tempo medio di approvazione in AIFA è stato di 248 giorni, contro i 441 giorni necessari per completare l’iter in EMA. A livello europeo non è stata riscontrata particolare differenza di tempistiche tra farmaci orfani e non orfani (450 vs 428 giorni), mentre in Italia la procedura di approvazione è risultata notevolmente più rapida per i farmaci non orfani (335 vs 197 giorni). Un dato interessante riguarda il confronto tra i periodi 2013-2014 e 2015-2016 che ha mostrato una diminuzione da 264 a 219 giorni nel tempo di approvazione in AIFA. A livello regionale i tempi di accesso variano notevolmente da Regione a Regione, passando dai 127 giorni necessari per l’accesso in Piemonte e Valle d’Aosta ai 279 giorni registrati in Basilicata.
Strumenti di Early Access
In Europa
Il Regolamento CE n. 726/2004, per l’istituzione di procedure comunitarie per l’autorizzazione e la sorveglianza dei medicinali per uso umano e veterinari, oltre a stabilire, come visto in precedenza, i criteri per l’accesso alla procedura di approvazione centralizzata, ha anche affermato la necessità di istituire «procedure di valutazione accelerate per i medicinali di maggiore interesse terapeutico e procedure per ottenere autorizzazioni temporanee soggette a condizioni annualmente verificabili». Per attuare le disposizioni del Regolamento 726/2004 l’EMA ha adottato diversi strumenti per accelerare le procedure di autorizzazione dei nuovi farmaci.
Accelerated Assessment
La procedura di valutazione accelerata riduce i tempi di valutazione della domanda di autorizzazione da 210 giorni a 150 o meno. La richiesta deve essere effettuata circa 2-3 mesi prima dell’applicazione della domanda ed è applicabile per i farmaci di maggior interesse per la salute pubblica in particolare sotto il punto di vista dell’innovatività.
Conditional Marketing Authorisation (CMA)
La richiesta di autorizzazione condizionata permette di ottenere l’AIC in tempi più brevi anche in assenza di dati clinici completi e con il vincolo di presentare dati a supporto per consolidare il profilo di efficacia e sicurezza. La Conditional Marketing Authorisation può essere richiesta per farmaci orfani, farmaci per trattare situazioni di emergenza o farmaci per il trattamento di malattie che mettono in pericolo la vita. In generale i dati a disposizione devono dimostrare che i benefici derivanti dalla disponibilità immediata del farmaco sono maggiori dei rischi correlati ai dati clinici mancanti. L’autorizzazione condizionata ha validità di 1 anno e può essere rinnovata annualmente. Può essere convertita in AIC permanente dopo presentazione dei dati mancanti.
Marketing Authorisation under Exceptional Circumstances (UEC)
L’AIC in circostanze eccezionali viene concessa ai quei farmaci per i quali non è possibile ottenere, neanche dopo l’autorizzazione, dati completi a supporto di efficacia e sicurezza in normali condizioni di utilizzo in quanto indicati per patologie molto rare o per ragioni etiche. Questa tipologia di autorizzazione ha validità per 5 anni ed è rinnovabile, ma il rapporto beneficio/rischio viene rivalutato annualmente dal CHMP e generalmente non viene convertita in AIC standard.
In Italia
In Italia sono stati introdotti diversi strumenti normativi per anticipare l’accesso nel mercato italiano di farmaci approvati in Europa.
Legge 648/1996
La Legge n. 648 del 23 dicembre 1996 prevede la possibilità di erogare, a carico del SSN e previo parere vincolante della CTS, farmaci per patologie per le quali non siano ancora disponibili alternative terapeutiche valide e che siano:
innovativi e già in commercio in altri Stati, ma non ancora autorizzati in Italia;
non ancora autorizzati e sottoposti a sperimentazione clinica;
disponibili in Italia, ma approvati con un’indicazione terapeutica diversa.
Inoltre, la Legge n. 79 del 2014 ha esteso l’applicazione di questa norma anche in presenza di alternative terapeutiche valide per medicinali da impiegare per un’indicazione terapeutica diversa da quella autorizzata purché tale indicazione sia nota e conforme a ricerche condotte nell’ambito della comunità medico-scientifica nazionale e internazionale, secondo parametri di economicità e appropriatezza.
Infine, il provvedimento della Commissione Unica del Farmaco del 20 luglio 2000 ha specificato la necessità di studi clinici di fase II affinché un farmaco possa essere inserito nell’elenco previsto dalla Legge 648/1996.
Fondo AIFA del 5%
La Legge n. 326 del 24 novembre 2003 ha istituito un fondo nazionale alimentato dalle aziende farmaceutiche con un contributo pari al 5% delle spese annualmente sostenute per le attività di promozione dirette al medico. Il 50% di tale fondo è destinato alle strutture sanitarie del SSN per l’acquisto di farmaci orfani o di farmaci non ancora autorizzati, ma che rappresentano una speranza di cura per gravi patologie. Le richieste di accesso al fondo fanno riferimento al singolo paziente e possono essere inoltrate dalle Regioni, dai centri di riferimento che hanno in cura i pazienti o da strutture specialistiche individuate dalle Regioni.
Criticità. Il Fondo AIFA 5% è attualmente poco utilizzato: nel 2017 sono stati erogati € 13,5 milioni su una disponibilità di circa € 17,8 milioni.
Nuove proposte. Dall’istituzione, nel 2003, del fondo AIFA 5% la ricerca medica ha fatto notevoli passi avanti e oggi sono disponibili nuove terapie per il trattamento di forme rare di tumore che, sebbene non siano ancora rimborsate, hanno a loro supporto robuste evidenze di efficacia. Una revisione della Legge 326/2003 dovrebbe aggiornare i criteri di accessi al fondo includendo, accanto ai farmaci orfani, anche i trattamenti per le forme rare di tumore [AIOM, 2018].
Utilizzo off-label
La Legge n. 94 del 8 aprile 1998 consente la prescrizione, da parte del medico che se ne assume esclusiva e diretta responsabilità, di un farmaco già commercio per l’uso al di fuori delle indicazioni terapeutiche o modalità di somministrazione per il quale è stato approvato. L’utilizzo off-label è rimborsato dalla Regione ed è consentito per il singolo paziente nel caso in cui si ritenga che non possa essere adeguatamente trattato coi farmaci a disposizione per quella determinata indicazione terapeutica.
Uso compassionevole
Il Decreto Ministeriale del 7 settembre 2017 stabilisce i criteri e le modalità per l’uso compassionevole di farmaci:
non ancora autorizzati in Italia, ma soggetti a sperimentazione clinica (in particolare, il farmaco deve essere oggetto di studi di fase II o III, o di fase I in caso di malattie rare e tumori rari. Inoltre, devono essere disponibili dati sufficienti per formulare un giudizio favorevole su efficacia e tollerabilità);
provvisti di AIC per indicazioni diverse;
autorizzati in altri paesi, ma non ancora in Italia.
Il farmaco può essere impiegato per uso compassionevole, e pertanto fornito a titolo gratuito da parte dell’azienda, per il trattamento di malattie rare, di pazienti gravi, in pericolo di vita, oppure affetti da malattie per le quali non esistano alternative terapeutiche valide.
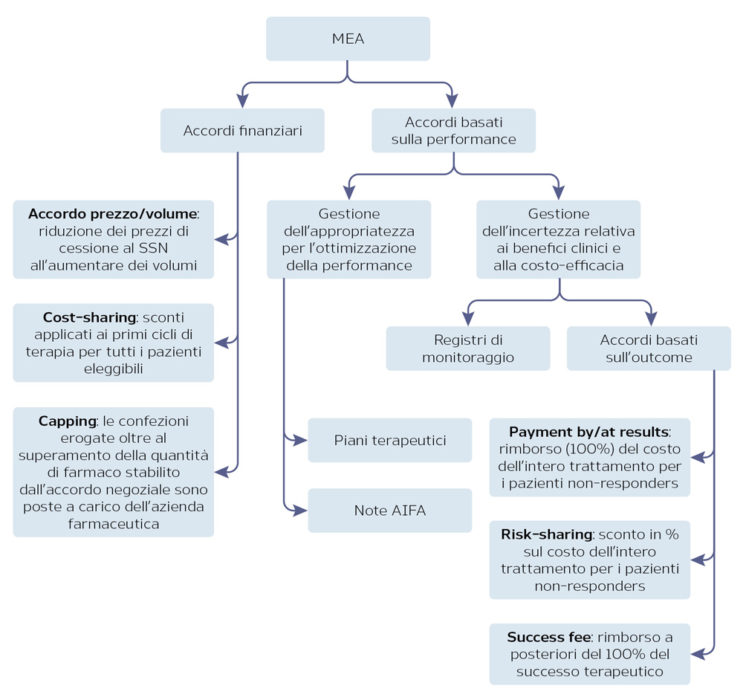
Managed Entry Agreements (MEA)
La necessità di accelerare i tempi di approvazione delle nuove terapie, al fine di permettere il rapido accesso sul mercato, si scontra con le esigenze dei servizi sanitari di contenere la spesa e di garantire l’efficacia e la sicurezza del trattamento. Gli strumenti per garantire l’appropriatezza terapeutica e la sostenibilità economica adottati da AIFA sono gli accordi di accesso condizionato (MEA) e i registri di monitoraggio a supporto di tali schemi. I registri di monitoraggio permettono di superare le incertezze derivanti dalla mancanza di evidenze del farmaco nel real-world, permettendo il monitoraggio dell’appropriatezza prescrittiva e la raccolta di dati epidemiologici e di sicurezza. I dati raccolti mediante il registro di monitoraggio permettono infine di gestire i MEA negoziati in fase di approvazione.
I MEA possono essere accordi di tipo finanziario o basati sulla performance del farmaco (Figura 2). In generale, oltre a permettere un più rapido accesso al mercato per i nuovi farmaci, i MEA consentono al SSN di condividere con l’azienda il rischio derivante dall’assenza di dati real-world e di limitare pertanto l’impatto sulla spesa farmaceutica.
Figura 2. I MEA in Italia
Fonte: AIFA
In merito all’utilizzo dei MEA, lo studio di Villa et al., nel quantificare i tempi di accesso ai farmaci, ha anche indagato l’impatto di strumenti di early access applicati a livello europeo (Conditional Marketing Authorisation – CMA e Marketing Authorisation under Exceptional Circumstances – UEC) sulla procedura di approvazione italiana, in particolare quanto l’incertezza derivante dal processo di autorizzazione a livello centrale abbia influenzato il processo di rimborsabilità a carico del SSN e il ricorso a MEA.
L’analisi ha considerato 65 farmaci che sono stati autorizzati in Europa con CMA (n. 35) o UEC (n. 30) tra luglio 2006 e febbraio 2017. Dei 35 farmaci approvati in Europa con CMA, 28 sono disponibili in Italia e 18 (64%) hanno ottenuto la rimborsabilità in classe A o H, mentre dei 30 autorizzati con UEC, 19 sono disponibili in Italia e 13 (68%) sono stati inseriti in classe A o H. L’analisi dei dati ha inoltre mostrato come l’approvazione a livello europeo mediante CMA sia predittiva di un successivo MEA al momento della negoziazione in AIFA: infatti, dei 18 farmaci approvati con CMA che hanno ottenuto la rimborsabilità in Italia, a 11 (61%) è stato applicato un MEA (5 cost-sharing, 5 payment by results e 1 risk sharing), mentre nessun MEA è stato applicato ai farmaci approvati con UEC.
Per quanto riguarda le tempistiche di approvazione, dal rilascio del parere della CHMP alla pubblicazione della determina di P&R, la procedura è più rapida per i farmaci approvati con CMA (636 vs 897 giorni), tuttavia considerando solo l’iter nazionale (dall’applicazione della domanda di P&R alla pubblicazione in GU) i farmaci autorizzati con UEC presentano tempi più rapidi (329 vs 510 giorni).
Infine, l’introduzione nell’elenco previsto dalla Legge 648/96, richiesto per 3 dei 18 farmaci approvati con CMA e per 6 dei 19 approvati con UEC, ha velocizzato la procedura di approvazione (195 vs 548 giorni per i farmaci CMA e 313 vs 713 per i farmaci UEC), ma ha rallentato l’iter di negoziazione di P&R (805 vs 548 giorni per i farmaci CMA e 818 vs 713 per i farmaci UEC).
Nel mondo
In Tabella I sono riportati alcuni esempi di schemi di early access applicati in USA e in diversi paesi europei.
Permette ai pazienti con malattie croniche o in pericolo di vita, per i quali non sono disponibili alternative terapeutiche, di accedere a un farmaco sperimentale al di fuori dagli studi clinici
Francia
Agence Nationale de Sécurité du Medicament et des Produits de Santé (ANSM) www.ansm.sante.fr
Autorisations Temporaires d’Utilisation (ATU)
Permette l’utilizzo di farmaci non ancora approvati e non soggetti a sperimentazione clinica per il trattamento di malattie gravi o rare per le quali non esiste un’alternativa terapeutica adeguata. Può essere di coorte se l’utilizzo è destinato a gruppi o sottogruppi di pazienti trattati e monitorati secondo criteri definiti in un protocollo terapeutico; sia la prescrizione che la fornitura sono limitate agli ospedali, che negoziano direttamente le condizioni iniziali di “tariffazione gratuita” con l’azienda e il rimborso completo è fornito dal sistema sanitario nazionale. L’ATU nominativo è riferito al singolo paziente e riguarda farmaci il cui rapporto efficacia/sicurezza è ritenuto favorevole alla specifica condizione
Germania
Das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) www.bfarm.de
Arzneimittel-Härtefall-Verordnung (AMHV)
Consente la distribuzione di farmaci non ancora autorizzati, con trattative di rimborso valutate caso per caso
UK
Medicines and Healthcare products Regulatory Agency (MHRA) www.gov.uk
Early access to medicines scheme (EAMS)
Permette l’accesso a farmaci non ancora autorizzati per il trattamento di condizioni potenzialmente letali o gravemente debilitanti in presenza di evidente unmet medical need. L’agenzia regolatoria valuta il rapporto benefici/rischi del farmaco che viene completamente rimborsato
Tabella I. Esempi di schemi di early access in altre nazioni
Alcune proposte di miglioramento dei programmi di Early Access in Italia
Farmaci orfani
Una serie di proposte per il miglioramento del quadro normativo che regola l’accesso anticipato ai farmaci orfani arriva da un Position Paper pubblicato nel 2018 dall’Osservatorio Farmaci Orfani (OSSFOR) e che ha identificato soluzioni a breve, medio e lungo termine per migliorare l’applicazione della normativa esistente (Legge 648/1996 e Legge 326/2003), ridurre i tempi di approvazione e permettere l’accesso da parte del paziente in tempi più rapidi. In generale, le principali criticità riscontrate riguardano la necessità di un dialogo precoce tra i soggetti coinvolti nei processi e maggiore condivisione delle informazioni.
Le proposte a breve termine comprendono l’agevolazione del confronto tra AIFA e aziende per la verifica della presenza dei presupposti per l’erogazione del farmaco stesso, l’istituzione di un registro delle autorizzazioni all’accesso al fondo AIFA 5% con elenco dei farmaci erogati e relative patologie e il ripristino del rimborso diretto di AIFA alle aziende. Nel medio periodo si propone la creazione di un registro di monitoraggio ad hoc per i dati clinici ed economici dei farmaci inseriti nell’elenco previsto dalla Legge 648/96 e la pubblicazione del procedimento seguito da AIFA per l’autorizzazione all’erogazione dei fondi AIFA 5%. Inoltre, sempre nel medio termine, potrebbero essere superate le criticità associate all’importazione dei farmaci mediante l’emanazione di indicazioni specifiche e linee guida da parte di AIFA.
La normativa italiana può contare su diversi strumenti di early access
Infine, le soluzioni di lungo termine, che riguardano interventi sul piano normativo, suggeriscono la modifica della Legge 648/96 per permettere il coinvolgimento delle aziende in fase di richiesta di inserimento nella lista, la disciplina dei prodotti a cui viene negato il rimborso, la predisposizione di un fondo di riferimento aggiornato annualmente, l’istituzione di un fondo dedicato ai farmaci off-label e la riduzione dell’incertezza sui tempi di negoziazione.
Farmaci oncologici
L’accesso tempestivo alle nuove terapie antitumorali con un significativo valore aggiunto rappresenta una necessità importante per i pazienti e con questo obiettivo un panel di esperti formato da medici oncologi, rappresentanti delle istituzioni, delle aziende farmaceutiche, delle associazioni di pazienti e dei media ha avanzato delle proposte per migliorare gli strumenti di early access a oggi disponibili in Italia [Apolone et al. 2019].
La prima proposta riguarda la definizione dei farmaci meritevoli di early access limitando l’accesso ai farmaci già valutati in EMA con procedura accelerata in quanto ritenuti innovativi e ai nuovi farmaci il cui medical need e valore aggiunto venga valutato secondo criteri ben definiti basati sui criteri di innovatività considerati da AIFA, sulle raccomandazioni all’uso dei farmaci elaborate dall’Associazione Italiana Oncologia Medica (AIOM) e sui criteri per la stratificazione del grado di beneficio clinico dei farmaci della European Society for Medical Oncology (ESMO).
I setting di applicazione di tali criteri sono quello curativo e non curativo, nel caso di malattia in stadio molto avanzato.
I farmaci che otterranno un punteggio superiore alla soglia definita verranno definiti first in class e potranno accedere al percorso di early access che prevede la possibilità di inoltrare la richiesta in AIFA subito dopo il parere positivo del CHMP. Il tempo concesso alla CTS per la valutazione del dossier e l’emissione del giudizio è di 60 giorni dopo il quale, in caso di parere positivo, il farmaco potrà entrare sul mercato ed essere completamente rimborsato dal SSN a un prezzo negoziato in via temporanea e con apposito registro di patologia. Dopo al massimo 12 mesi, al termine della procedura di P&R l’azienda dovrà restituire l’eventuale differenza tra il prezzo definito per l’early access e quello infine negoziato. In caso di rimborsabilità negata il farmaco perde la rimborsabilità e, mentre ai pazienti verrà garantita la continuità terapeutica, l’azienda dovrà rimborsare la spesa sostenuta dal SSN.
I farmaci che, secondo il punteggio di valore clinico, verranno definiti second in class potranno accedere al percorso di early access solo in presenza di evidenze scientifiche che ne dimostrino il beneficio clinico aggiuntivo.
Infine, secondo il panel di esperti l’early access dei farmaci oncologici potrebbe essere finanziato dalla quota non erogata del fondo AIFA 5%, dai rimborsi che l’azienda dovrà eventualmente versare come differenza di prezzo e dal pay back della spesa sostenuta in caso di non rimborsabilità. Nel caso in cui al farmaco venga riconosciuta l’innovatività, il percorso di early access si fermerebbe e il farmaco potrà accedere ai fondi previsti per i farmaci oncologici innovativi.
Bibliografia di riferimento
Apolone G, Ardizzoni A, Buzzetti G, et al. Early Access in Oncology: why is it needed? Global & Regional Health Technology Assessment 2019; 2019: 1-7
Associazione Italiana di Oncologia Medica – AIOM. Early access in Italia. Le regole che definiscono l’accesso anticipato ai farmaci anticancro. AIOM, 2018
Decreto 7 settembre 2017. Disciplina dell’uso terapeutico di medicinale sottoposto a sperimentazione clinica. GU Serie Generale n.256 del 02-11-2017
Legge 23 dicembre 1996, n. 648. Conversione in legge del decreto-legge 21 ottobre 1996, n. 536, recante misure per il contenimento della spesa farmaceutica e la rideterminazione del tetto di spesa per l’anno 1996. GU Serie Generale n.300 del 23-12-1996
Legge 24 novembre 2003, n. 326. Conversione in legge, con modificazioni, del decreto-legge 30 settembre 2003, n. 269, recante disposizioni urgenti per favorire lo sviluppo e per la correzione dell’andamento dei conti pubblici. GU Serie Generale n.274 del 25-11-2003 – Suppl. Ordinario n. 181
Legge 8 aprile 1998, n. 94. Conversione in legge, con modificazioni, del decreto-legge 17 febbraio 1998, n. 23, recante disposizioni urgenti in materia di sperimentazioni cliniche in campo oncologico e altre misure in materia sanitaria. GU Serie Generale n.86 del 14-04-1998
Legge 8 novembre 2012, n. 189 . Conversione in legge, con modificazioni, del decreto-legge 13 settembre 2012, n. 158, recante disposizioni urgenti per promuovere lo sviluppo del Paese mediante un più alto livello di tutela della salute. GU Serie Generale n.263 del 10-11-2012 – Suppl. Ordinario n. 201
Lidonnici D, Ronco V, Isernia M, et al. Tempi di accesso ai farmaci in Italia nel periodo 2015-2017: Analisi delle tempistiche di valutazione dell’Agenzia Italiana del Farmaco. Global & Regional Health Technology Assessment 2018; 2018: 1-9
Prada M, Ruggeri M, Sansone C. Timeline of Authorization and Reimbursement for Oncology Drugs in Italy in the Last 3 Years. Medicine Access @ Point of Care 2017; 1: e29-36
Provvedimento Commissione Unica del Farmaco 20 luglio 2000. Istituzione dell’elenco delle specialità medicinali erogabili a totale carico del Servizio sanitario nazionale ai sensi della legge 648/96. GU n. 219 del 19-09-2000
Regolamento (CE) n. 726/2004 del Parlamento europeo e del Consiglio del 31 marzo 2004 che istituisce procedure comunitarie per l’autorizzazione e la sorveglianza dei medicinali per uso umano e veterinario, e che istituisce l’agenzia europea per i medicinali. Gazzetta ufficiale n. L 136 del 30/04/2004. Disponibile su https://eur-lex.europa.eu/legal-content/IT/TXT/HTML/?uri=CELEX:32004R0726&from=IT (ultimo accesso settembre 2019)
Villa F, Jommi C, Genazzani A, et al. Accesso precoce al mercato: dalle approvazioni condizionate di EMA agli accordi negoziali particolari di AIFA. Global & Regional Health Technology Assessment 2018; 2018: 1-10
Il punto sulla risoluzione del Ministero della Salute
Il Ministero della Salute ha promosso presso l’Organizzazione Mondiale della Sanità una risoluzione sul tema della trasparenza sul mercato dei farmaci, dei vaccini e delle altre tecnologie sanitarie, supportata da alcuni altri stati, tra i quali al momento non compaiono i principali Paesi Europei, con l’eccezione della Spagna. Il documento contiene diverse riflessioni, dalla disponibilità di dati clinici, alle informazioni sulla scadenza dei brevetti, ai costi sostenuti dalle imprese, ai prezzi dei farmaci e si inquadra nelle iniziative che l’OMS ha promosso per aumentare l’equità nell’accesso a farmaci e tecnologie sanitarie, incluso il Fair Price Forum.
Su questo documento, i ricercatori dell’Osservatorio Farmaci del Cergas Bocconi hanno già espresso una valutazione critica e articolata in un articolo comparso sul Sole 24 Ore Sanità [1], in cui sono stati rilevati:
la necessità di focalizzarsi sulle criticità delle attuali stime sui costi di ricerca e sviluppo e produzione di farmaci, invece di richiedere alle imprese valutazioni puntuali su tali costi nella negoziazione dei prezzi, che dovrebbe invece basarsi sul valore aggiunto dei farmaci per il sistema sanitario;
il rischio che la trasparenza piena dei prezzi, ovvero la visibilità di sconti ed effetti di altri accordi negoziali che hanno garantito l’accessibiltà dei farmaci in paesi con diversa capacità (e disponibilità) di pagare, possa generare ritardi importanti nel lancio dei prodotti proprio sui mercati che hanno beneficiato maggiormente di tali accordi, per effetto di pratiche diffuse da parte dei pagatori di cross-reference pricing (ovvero di fissazione dei prezzi domestici sulla base dei prezzi negoziati in altri paesi);
l’opportunità che la trasparenza (e l’apertura al mondo degli stakeholder) venga evocata anche sui processi decisionali e negoziali di valutazione, rimborso e prezzo, sia in termini di esplicitazione più dettagliata ex ante dei criteri decisionali, sia in termini di reportistica consultabile da tutti in merito alle valutazioni e agli esiti dei processi decisionali ex post.
Prezzo dei farmaci: ma è tutto così invisibile?
La visibilità di sconti ed effetti di altri accordi negoziali potrebbe rappresentare un ostacolo al lancio di nuovi farmaci in determinati mercati
Il dibattito collegato alla trasparenza dei prezzi ha assunto che le informazioni sui prezzi e sugli accordi che generano un differenziale tra prezzo negoziato (o liberamente determinato dalle imprese) e prezzo effettivo (al netto di sconti e/o degli effetti di MEA – Managed Entry Agreements) non siano disponibili. Ciò è solo parzialmente corretto.
Se si prendono i principali Paesi Europei (Francia, Germania, Italia, Spagna e Regno Unito), in tutti i paesi i prezzi negoziati (Francia, Italia e Spagna) o liberamente determinati dalle imprese (Germania e Regno Unito) sono noti. In tutti questi paesi sono stati però introdotti sconti sul prezzo e/o MEA, finanziari (tetti complessivi sulle vendite / spesa per farmaci, accordi prezzo/volume, capping sulla spesa per prodotto, capping sul costo per prodotto per paziente trattato, copertura da parte delle imprese dei primi cicli di terapia) e outcome-based (payback da parte delle imprese in caso di mancata risposta del paziente – cosiddetti Payment by Result – PbR; rinegoziazione dei prezzi sulla base di evidenze in real-life su popolazione trattata – cosiddetti Coverage with Evidence Development – CED). La Tabella I illustra la diffusione di tali accordi nei suddetti paesi distinguendo tra iniziative a livello centrale, (eventualmente) regionale e locale, ovvero di singole aziende sanitarie [2].
Centrale
Accordi prezzo/volume
Sconti
Alcuni CED
Sconti (AMNOG)
Tutti tranne i CED
Sconti
Cap ai costi
Target sulle vendite (PPRS)
Sconti
Alcuni altri
Regionale
–
Sconti
Sconti
Sconti
Accordi prezzo/volume
PbR
–
Locale
–
Sconti
Sconti
Sconti
Accordi prezzo/volume
PbR
Sconti
Tabella I. Sconti / MEA nei principali Paesi Europei
AMNOG = Arzneimittelmarktneuordnungs-gesetz (regolazione del mercato farmaceutico introdotta nel 2011); CED = Coverage with Evidence Development; PbR = Payment by Results; PPRS = Pharmaceutical Price Regulation Scheme
Applicazione sconto e/o MEA su singolo farmaco
Solo CED
AMNOG
Impatto finanziario sconto e/o MEA aggregato
Solo per accordi prezzo/volume (trattenuto da assicurazioni sociali)
Se gestiti attraverso payback
Solo per PPRS
Impatto finanziario sconto e/o MEA per prodotto
AMNOG
Solo payback su tetti per prodotto e sconti per tutti i prodotti
Solo payback su tetti per prodotto e sconti per tutti i prodotti
Solo per PPRS
Sconto in acquisti
–
Tabella II. Le informazioni disponibili su sconti / MEA
AMNOG = Arzneimittelmarktneuordnungs-gesetz (regolazione del mercato farmaceutico introdotta nel 2011); CED = Coverage with Evidence Development; PPRS = Pharmaceutical Price Regulation Scheme
Il Fair Pricing Group dell’OMS ha considerato come primo step desiderabile la semplice segnalazione dell’esistenza di un accordo negoziale (essendo il suo contenuto confidenziale): il dato sull’esistenza degli specifici accordi è disponibile in Italia, Germania e UK mentre quello sul loro impatto è abbastanza limitato (Tabella II).
Nello specifico, in Francia, dove è molto frequente la stipulazione di accordi prezzo/volume, non è noto quali prodotti siano soggetti a tale accordo, anche se ne è noto l’effetto finale finanzario aggregato (tale importo viene pubblicato ogni anno sul Rapport d’Activite del CEPS – Comité économique des produits de santé).
La Germania è l’unico paese a pubblicare gli sconti negoziati sui prezzi liberamente determinati dalle imprese ad un anno dal lancio del prodotto, ma non è noto quale sia l’effetto finanziario complessivo di tali accordi.
In UK viene regolarmente pubblicata la lista dei farmaci soggetti a Patient Access Scheme / Commercial Agreements (in prevalenza si tratta di sconti), ma l’entità di tali sconti non è nota, mentre è solo (parzialmente) conosciuto l’effetto aggregato dei payback associati al superamento dei target di vendite negoziati dalle imprese.
In Spagna le informazioni sono molto limitate.
Paradossalmente l’Italia è il paese in cui le informazioni sono maggiori. È noto, in quanto pubblicato in Gazzetta Ufficiale, che tipo di accordo è stato stipulato con AIFA (sconto / MEA per tipologia).
In Italia le informazioni sul prezzo dei farmaci sono maggiormente accessibili rispetto a Francia, Spagna, Germania e UK
Gli sconti confidenziali (a livello nazionale) sono noti a tutti coloro che acquistano i farmaci (il prezzo di partenza per l’acquisto è quello al netto dello sconto confidenziale negoziato con l’AIFA), ma non ne si conosce l’impatto finanziario aggregato. I bandi di gara (o le eventuali trattative negoziali pubblicate in Gazzetta Ufficiale) rendono però parzialmente visibili anche al pubblico generale gli effetti di tali accordi: nel bando viene, infatti, esplicitato il prezzo base d’asta che è, al più, pari al prezzo al netto degli sconti confidenziali stipulati con l’AIFA. Dalla stessa fonte è possibile risalire anche agli sconti negoziati a livello regionale / locale. I MEA sono accessibili (con alcune eccezioni: non lo sono stati, ad esempio, in fase iniziale gli accordi prezzo/volume collegati ai farmaci per Epatite C) a clinici e farmacisti che gestiscono i registri farmaci. È infine noto l’effetto dei MEA a livello finanziario aggregato: il dato viene pubblicato sui documenti AIFA di monitoraggio della spesa per farmaci. L’Osservatorio Farmaci del Cergas, attraverso dati pubblici, ha stimato, per i farmaci acquistati dalle aziende sanitarie / regioni per il 2017, un differenziale aggregato del 37% tra valore di acquisto al prezzo pubblicato in Gazzetta Ufficiale e costo effettivo per il SSN, di cui il 25% come effetto di sconti nazionali ed extra-sconti in sede di gara, il 10,3% come effetto di MEA finanziari e l’1,7% come effetto del payback associato ad accordi outcome-based [3].
La trasparenza sui processi e sull’esito
La valutazione dei farmaci (assessment) e le decisioni sul rimborso e prezzo (appraisal) sono molto complesse e delicate ed è giusto che venga mantenuto un certo margine di confidenzialità e discrezionalità. La trasparenza e, soprattutto, la percezione di equità nell’applicazione di criteri e processi negoziali sono però molto importanti, soprattutto laddove l’esito di una decisione può impattare sullo stato di salute dei pazienti. In questo senso la trasparenza (e riproducibilità delle decisioni) evocata sul tema dei prezzi andrebbe soprattutto applicata al processo di valutazione e decisione di rimborso e prezzo (Tabella III).
Documenti su assessment benefici
SMR/ASMR
Beneficio incrementale
Solo per valutazione innovatività
Solo per indicazione terapeutica rimborsata
∆ QALYs
Documenti su appraisal benefici
SMR/ASMR
Beneficio incrementale
Solo per valutazione innovatività
Solo per indicazione terapeutica rimborsata
∆ QALYs
Documenti su assessment economico
RICE (Avis d’efficiencie) se richiesto
RICE (Appraisal document)
Negoziazione dei prezzi
–
–
Definizione tempistica ex ante
∼
Solo 100 giorni per AIC ricondotta a P&R
Pubblicazione scadenze Appraisal Document
Dato su durata del processo
Dato aggregato
Tabella III. Livello di trasparenza del processo
ASMR = Amélioration du Service Médical Rendu (valore terapeutico aggiunto);
SMR = Service Médical Rendu (valore terapeutico);
RICE = Rapporto Incrementale di Costo-Efficacia;
AIC = Autorizzazione all’Immissone in Commercio;
P&R = Prezzo e Rimborso;
QALYs = Quality Adjusted Life Years (saved)
In un’ottica di equità dei criteri negoziali, il processo di valutazione e decisione di rimborso e prezzo andrebbe reso ancora più trasparente
Su questo aspetto l’Italia ha fatto grandi passi in avanti con la pubblicazione delle schede di valutazione dell’innovatività. Tuttavia, il nostro paese presenta ancora un livello piuttosto basso di trasparenza non solo nella fase di negoziazione dei prezzi strictu sensu (aspetto che lo accomuna agli altri Paesi Europei in cui il prezzo viene negoziato), ma anche nella definizione, ex ante rispetto alla negoziazione ed ex post rispetto alle decisioni prese, degli aspetti chiave del processo negoziale, ovvero quale comparatore sia stato scelto per la valutazione del beneficio relativo, quanto sia stato rilevante il beneficio incrementale per ottenere un premio di prezzo, quali siano i domini del beneficio più rilevanti, quanto le eventuali evidenze di costo-efficacia e budget impact siano state considerate per decidere se il farmaco offrisse, rispettivamente, un beneficio coerente con il prezzo richiesto e un impatto regionevole sulla spesa.
Con questo non si vuole affermare che gli altri sistemi siano del tutto trasparenti: in Francia, ad esempio, non è noto quanto il beneficio incrementale si sia tradotto in un premio di prezzo o quanto invece il prezzo sia stato negoziato sulla base dell’impatto sulla spesa, ma alle imprese è noto che solo con un beneficio incrementale da moderato a importante è possibile aspirare ad un premio di prezzo.
In Germania non viene esplicitato ex post quali siano stati i fattori effettivamente impattanti sullo sconto rispetto al prezzo liberamente determinato dalle imprese, ma esiste un’evidenza abbastanza consolidata di letteratura che mostra come il beneficio incrementale sia uno dei fattori determinanti lo sconto [4] e il differenziale di prezzo rispetto al farmaco considerato come comparatore nell’ambito del sistema AMNOG [5].
I passi in avanti da fare per una maggiore trasparenza a tutto vantaggio del processo negoziale in Italia sono però ancora molti.
Bibliografia
Jommi C, Armeni P, Costa F, et al. Trasparenza del prezzo dei farmaci: il punto sulla lettera della salute all’OMS. Il Sole 24 ore Sanità. 2 aprile 2019
Jommi C. Managed entry agreements and high cost medicines (European perspective). In: Zaheer-Ud-Din Babar (ed). Equitable access to high-cost pharmaceuticals. London: Springer, 2018
Nell’ultima sessione plenaria prima delle elezioni europee, ad aprile scorso, il Parlamento Europeo ha approvato, a larga maggioranza (572 voti a favore, 36 contro, 22 astenuti), il Regolamento europeo di deroga al certificato protettivo supplementare (Supplementary protection certificate – SPC – manufacturing waiver). Il Regolamento è successivamente stato approvato anche dal Consiglio Europeo e dovrebbe essere pubblicato sulla Gazzetta Ufficiale dell’Unione Europea prima dell’estate.
Come sottolinea Assogenerici, per la prima volta l’Unione Europea è intervenuta in materia di proprietà intellettuale per promuovere la competitività dell’industria manifatturiera con sede negli stati della UE e, in particolare, delle aziende produttrici di generici e biosimilari.
Ma di che cosa si tratta e quali sono i benefici auspicati per l’industria europea del farmaco? Il certificato protettivo supplementare (SPC), introdotto nel 1992, riguarda l’estensione di 5 anni della copertura brevettuale riservata ai farmaci, che in Europa dura 20 anni. Il testo finale approvato dal Parlamento e dal Consiglio UE, che emenda il precedente Regolamento UE n. 469/2009, prevede che le aziende con sede in UE possano avviare la produzione di un medicinale generico o biosimilare in deroga all’SPC, e quindi durante gli ulteriori 5 anni di copertura brevettuale, in due casi specifici:
per l’export verso un Paese extra-UE dove il farmaco non sia coperto da brevetto;
per stoccarlo negli ultimi 6 mesi di validità dell’SPC e immetterlo in commercio il giorno dopo la scadenza brevettuale in UE (il cosiddetto “Day-1 Launch”).
L’SPC manufacturing waiver è previsto anche in Usa, Canada e Giappone. Per i primi 3 anni la deroga si applicherà agli SPC richiesti dopo l’entrata in vigore del Regolamento, quindi a partire da luglio 2022.
Il Regolamento prevede una serie di misure di salvaguardia, come l’obbligo per l’azienda che intenda avvalersi della deroga di informare il titolare del certificato, l’autorità competente e tutti gli attori della filiera, e l’obbligo di etichettatura dei farmaci destinati all’esportazione extra-UE apponendo un logo apposito (Figura 1).
Logo EU export
Figura 1. Logo che identifica i farmaci destinati ai paesi extra-UE prodotti con deroga all’SPC
Il nuovo Regolamento è frutto di un lungo lavoro di analisi della Commissione Europea che, tramite studi ad hoc, ha valutato le esigenze del settore soprattutto in relazione ai cambiamenti del mercato degli ultimi anni e ai possibili benefici di una deroga all’SPC, a partire dalla considerazione di quelle che vengono definite, dalla Commissione stessa, “conseguenze indesiderate” dell’estensione della copertura brevettuale sulla competitività delle aziende manifatturiere europee: nel periodo di validità dell’SPC le aziende europee non potevano, infatti, fabbricare generici e biosimilari né per l’esportazione in paesi terzi che non prevedevano la protezione legale né per stoccarli in sufficiente anticipo da poter essere immessi sul mercato dal primo giorno della scadenza brevettuale in UE.
Studi della Commissione hanno evidenziato come la presenza dell’SPC, nato per sostenere il comparto farmaceutico europeo, abbia invece contribuito all’aumento della delocalizzazione della produzione di generici e biosimilari in paesi extra-UE, con un impatto negativo generalizzato sull’economia dell’Unione.
Contrastare tale delocalizzazione e incentivare il ritorno di attività di ricerca, sviluppo e produzione in UE sono tra i principali obiettivi della Commissione. In termini di benefici economici, le stime pubblicate parlano di un aumento delle vendite nette di € 9,5 miliardi a seguito della possibilità di export verso paesi extra-UE, con la creazione di 25.000 nuovi posti di lavoro. Inoltre, l’aumento della concorrenza nell’Unione, come conseguenza di un più rapido accesso al mercato per generici e biosimilari, comporterebbe risparmi per la spesa farmaceutica per oltre 3 miliardi di euro.
Positivo il commento di Assogenerici al nuovo Regolamento, che in un comunicato del presidente, Enrique Häusermann, sottolinea: «La norma sull’SPC manufacturing waiver rende plausibile il nostro auspicio di poter aumentare fino al 70% la quota di fatturato derivante dalle nostre esportazioni, che attualmente si indirizzano soprattutto verso l’Europa e il Nord America e – in quota minore – verso l’Asia Orientale. In un’ottica di comparto allargato va anche ribadito che si tratta senz’altro di una strategia win-win: non si intacca in alcun modo la tutela garantita al titolare dell’SPC e si opera a vantaggio soprattutto delle piccole e medie imprese, che potranno competere sempre più ad armi pari con le aziende non europee, tornando ad essere nuovamente dei player strategici anche per le aziende multinazionali».
Forti perplessità invece dall’European Federation of Pharmaceutical Industries and Associations (EFPIA) che in una nota sottolinea il rischio di indebolimento della protezione della proprietà intellettuale e l’importante compito che spetta alla prossima Commissione Europea, alla quale chiede di «ristabilire l’equilibrio, sostenendo più in generale la ricerca, lo sviluppo e l’innovazione». Tra le richieste di EFPIA, il miglioramento dei percorsi di fast-track dedicati alle terapie innovative in grado di rispondere ad unmet needs, il mantenimento e il potenziamento di incentivi per la Ricerca & Sviluppo di nuove terapie dedicate a patologie con bisogni di salute insoddisfatti e il sostegno verso il Partenariato Pubblico Privato.
Fonti
Regolamento del Parlamento Europeo e del Consiglio che modifica il Regolamento (CE) n. 469/2009 sul certificato protettivo complementare per i medicinali. PE-CONS 52/1/19, 20 maggio 2019
Alcuni (dis)orientamenti giurisprudenziali recenti
L’importanza del settore dei farmaci biologici spiega perché, nonostante molti anni siano passati dall’introduzione dei primi farmaci biosimilari e dopo che faticosamente, anche per l’assenza di norme specifiche, la giurisprudenza abbia trovato punti di equilibrio funzionali e funzionanti per il sistema, i casi giudiziali che coinvolgono il rapporto tra originatori e biosimilari siano sempre molto presenti nella giurisprudenza amministrativa.
Talvolta, tuttavia, si ha l’impressione che ci si dimentichi dell’intero percorso evolutivo che la giurisprudenza, e la legge sulla scorta di essa, hanno compiuto negli ultimi dieci anni. Succede cioè che l’attenzione del giudice si concentri su aspetti di dettaglio della biosimilarità che, seppur effettivamente importanti e meritevoli di approfondimento individuale, tuttavia non possono essere esaminati senza tenere in considerazione l’intero quadro normativo, scientifico e – anche – giurisprudenziale in questa materia.
In particolare, due recenti decisioni dimostrano come in alcuni casi si finisca facilmente per complicare situazioni che, al contrario, dovrebbero ormai essere molto semplici.
Il caso che ha portato alla decisione del T.A.R. per la Toscana, Sez. II, n. 400 del 21 marzo 2019
Notevole è stato il risalto mediatico che la recente sentenza del T.A.R. per la Toscana, sez. II, n. 400 del 21 marzo 2019, ha avuto sulla stampa specializzata, che l’ha perlopiù enfatizzata per le posizioni di chiusura rispetto al biosimilare rispetto all’originatore (mi riferisco specialmente all’articolo apparso sul quotidiano Sanità24 del 26 marzo 2019, nel quale peraltro è riportato un numero della sentenza errato, dall’eloquente titolo “Biologici o biosimilari, sui farmaci va salvaguardata l’autonomia del medico”).
Il T.A.R. di Firenze, in effetti, non ha affatto chiuso ai biosimilari né affermato che tra biosimilare ed originatore il medico è libero di decidere in completa autonomia. Questa posizione sarebbe semplicemente in contrasto con le norme e con i principi giurisprudenziali tuttora in vigore.
Metodologicamente, l’esame della sentenza in questione non può prescindere dal punto di partenza essenziale: il contenuto del provvedimento impugnato. E qui deve essere rimarcato come, sorprendentemente, la delibera della Regione Toscana n. 194 del 2018 aveva previsto modalità di prescrizione, dispensazione e acquisizione di farmaci senza distinguere tra farmaci chimici (e relativi generici) e farmaci biotecnologici (e relativi biosimilari). Si erano previste, cioè, regole amministrative uniche, senza prevedere alcuna specificità per i farmaci biotecnologici. E correttamente il T.A.R. dà atto che questa è la premessa del proprio ragionamento, quando riconosce che la delibera impugnata è riferita ai farmaci in generale e non contiene alcuna regolamentazione specifica sui farmaci biologici e che, di conseguenza, risulta applicabile anche a questi ultimi.
In particolare, l’ente regionale aveva stabilito una procedura di controllo, da parte della Regione stessa, su eventuali richieste di acquisto di farmaci diversi da quelli aggiudicati inserite nel sistema informativo regionale, e che tale richiesta sarebbe stata esaminata in base, esclusivamente, alle politiche di governance della spesa e della sostenibilità dell’assistenza farmaceutica regionale.
Di fronte ad un contenuto di questo tipo è francamente inimmaginabile un esito diverso da quello dell’annullamento. Infatti, l’errore commesso dalla Regione è evidente sotto un duplice profilo: a) non avere previsto regole specifiche per i farmaci biologici; b) non avere tenuto conto che nel rapporto tra originator e biosimilari vi sono una serie di norme (art. 15 d.l. n. 95/2012 nel testo oggi vigente) e di regole (il secondo position paper di AIFA, in particolare) che prevedono un controllo medico sulla prescrizione.
È del tutto condivisibile, in questa ottica, che il T.A.R. per la Toscana abbia rimarcato alcuni principi imprescindibili quando si ha a che fare con i biosimilari.
Primo: ai farmaci biologici non si applica l’art. 7 d.l. n. 347/01 conv. in l. n. 405/01 che consente, per i soli farmaci chimici, la c.d. sostituibilità secondaria, ossia quella automatica, operata dal farmacista che fornisce al paziente il farmaco generico in assenza di specifica indicazione del medico prescrittore circa la non sostituibilità del farmaco prescritto. Per i biosimilari, tale sostituibilità secondaria, o automatica, è esclusa dall’art. 15, comma 11 quater, d.l. n. 95/12 conv. in l. n. 135/2012 ed introdotto dalla legge n. 232/16 che prevede sempre l’esclusivo controllo del medico sulla prescrizione.
Secondo: per i farmaci biologici non è consentito effettuare una gara ponendo nel medesimo lotto principi attivi differenti quando vi siano anche biosimilari (art. 15, comma 11 quater, comma 3, primo periodo, d.l. n. 95/12).
Terzo: le procedure di acquisto – cui anche la delibera regionale si riferiva – sono diverse, dal momento che quando i biosimilari siano almeno tre è obbligatoria la procedura di accordo quadro con tre aggiudicatari (art. 15, comma 11 quater, comma 3, lett. b).
Quarto: il medico che prescriva un farmaco biologico aggiudicato con questa particolare forma di procedura obbligatoria di accordo quadro con più operatori è libero di prescrivere uno dei primi tre farmaci, se ciò è preordinato a garantire la continuità terapeutica del paziente.
Libertà prescrittiva e governance della spesa sono due facce della stessa medaglia, non elementi contrapposti
La sentenza avrebbe potuto concludersi qui: questi quattro punti mostrano l’illegittimità del provvedimento della Regione, per non avere previsto queste peculiarità – peraltro discendenti direttamente dalla legge – per i farmaci biologici e biosimilari.
Ma è in questo contesto che va valutata l’ulteriore affermazione, quasi fosse un obiter dictum nella sentenza, sulla quale successivamente si sono concentrati tutti i commenti pro-originatore. Il Collegio infatti aggiunge che “se è ben possibile che la Regione eserciti, nell’ambito di politiche di razionalizzazione della spesa farmaceutica, una funzione di orientamento nei confronti dei medici prescrittori, permane tuttavia un “limite invalicabile” che è dato dall’autonomia decisionale del medico nella prescrizione di un farmaco, sotto il profilo dell’appropriatezza terapeutica”.
Il rapporto tra costo, costo-beneficio e libertà prescrittiva è infatti da tempo solcato dalla giurisprudenza amministrativa, che pare in effetti avere trovato un punto di equilibrio convincente. Il Consiglio di Stato ha da qualche anno precisato che “costituisce dovere, anche per il medico prescrittore quello di scegliere, tra prodotti ritenuti di pari efficacia, quello meno oneroso per il servizio sanitario nazionale” (Sez. III, sent. n. 5705 del 17 dicembre 2015 e n. 5776 del 18 dicembre 2015). Il concetto è stato ribadito anche successivamente (Sez. III, sent. n. 3621 del 21 luglio 2017) proprio in riferimento a provvedimenti regionali, valutati legittimi da Palazzo Spada, che prevedevano conseguenze a carico dei medici prescrittori che non tenessero in considerazione, all’atto della prescrizione, anche le valutazioni di costo della terapia unitamente alle esigenze terapeutiche, motivando specificamente perché intendessero discostarsi dalla prescrizione del farmaco a minor costo (per intolleranze, per continuità terapeutica, ecc.).
La più recente posizione del Consiglio di Stato, citata dalla stessa sentenza toscana, ha ulteriormente meglio precisato i rapporti esistenti tra le esigenze di contenimento della spesa farmaceutica regionale e la libertà prescrittiva del medico, ritenendo legittima e valorizzando “l’espressa previsione (…) che – qualora il medico ritenesse di non poter utilizzare il farmaco biosimilare o biologico originator al costo di terapia più basso rispetto al costo di terapia di altro farmaco biosimilare o biologico originator – fosse tenuto a motivare la scelta terapeutica, con specifica relazione indirizzata alla direzione sanitaria di appartenenza e di competenza territoriale dell’assistito” (Sez. III, n. 2821 del 11 maggio 2018). Cosicché, qualora tali cautele manchino, imponendo per esempio ai medici una rigida soglia minima di prescrizione di farmaco biosimilare o comunque a minor costo, i provvedimenti sarebbero al contrario illegittimi per violazione della libertà prescrittiva del medico.
Ecco, allora, che anche la sentenza del T.A.R. per la Toscana, pur nelle conclusioni del tutto corretta e condivisibile, appare forse un po’ troppo superficiale nell’affermare che la libertà prescrittiva è “limite invalicabile” e che le politiche di governance della spesa farmaceutica regionale non avrebbero alcun rilievo nella “valutazione medica di appropriatezza di cura e di garanzia della continuità terapeutica”.
La libertà prescrittiva deve essere garantita, certo: ma non è arbitrio prescrittivo. Deve essere esercitata dal medico all’interno del quadro di regole e di norme esistenti, tra cui l’indicazione (giurisprudenziale ma anche contenuta nell’art. 13 del codice di deontologia medica) secondo cui il costo della terapia è uno degli elementi che concorre, assieme alle valutazioni terapeutiche e cliniche, nella scelta del farmaco da prescrivere. Con la conseguenza che, a parità di effetti terapeutici tra biosimilare ed originator va preferito il farmaco che costa meno. Occorre solo precisare che questa parità di effetti va valutata nel caso concreto, ossia sul singolo paziente da parte del singolo medico; ciò dal momento che ex ante ed in astratto la parità di effetti terapeutici è ontologicamente insita nel concetto e nella definizione di farmaco biosimilare, nel quale le differenze con l’originator, pur esistenti ed anzi ineliminabili, sono già state valutate da EMA all’atto dell’autorizzazione all’immissione in commercio come non significative ai fini della conclusione che i due farmaci hanno lo stesso profilo di efficacia, sicurezza e d’uso.
Volendo sintetizzare: spesa farmaceutica e appropriatezza sono due facce della stessa medaglia. La prima non può prevalere sulla seconda. Ma neppure il contrario.
Lo strano caso dell’epoetina zeta
In questo contesto, di apparente scarsa attenzione sul valore scientifico del concetto di biosimilarità, pare interessante commentare brevemente anche la decisione, questa sì scarsamente condivisibile anche nelle conclusioni, con la quale il Consiglio di Stato ha ritenuto che l’epoetina zeta non sia un biosimilare dell’epoetina alfa (si tratta della sentenza della Sez. III, n. 871 del 5 febbraio 2019).
La sentenza infatti muove correttamente dall’esame dell’autorizzazione EMA dell’epoetina zeta, riconoscendo che effettivamente il farmaco è autorizzato come biosimilare e che il medicinale di riferimento è l’epoetina alfa (originatore, ovviamente). Tanto basterebbe per rispondere al quesito giuridico in senso diametralmente opposto rispetto a quanto affermato dalla sentenza stessa.
Sennonché, il ragionamento del Collegio si è concentrato su di una relazione istruttoria, acquisita al fascicolo, commissionata in primo grado con la quale era stato chiesto ad AIFA di chiarire cosa fosse l’epoetina zeta.
Ed in quella relazione, inevitabilmente, AIFA ha spiegato che il concetto di farmaco biosimilare ammette, ed anzi – aggiungo – non può prescindere, dal riconoscimento delle diversità esistenti rispetto al farmaco originator. Ciò è vero per qualsiasi tipo di farmaco biologico, ed è vero peraltro anche tra lotti produttivi diversi dello stesso originator. Come ben noto, questa diversità dipende dal fatto che nel farmaco biotecnologico il prodotto (a suo tempo coperto da brevetto) non è la molecola che si ottiene dalla cellula, ma il processo di produzione che genera quella molecola. Ripetendo il processo produttivo due, tre, dieci, mille volte, il risultato non sarà mai identico pur trattandosi dello stesso farmaco.
Eppure, nella sentenza il Consiglio di Stato afferma che le differenze nel modello di glicosilazione sono sufficienti per escludere l’identità del principio attivo, ancorché il farmaco sia stato autorizzato come biosimilare (perché avente lo stesso profilo di sicurezza ed efficacia terapeutica. Si badi: lo “stesso profilo”, e non solo un “profilo equivalente”).
Avremmo cioè un tertium genus, che va ad aggiungersi a biosimilare, e ad originator diverso ma equivalente: lo strano caso del biosimilare avente principio attivo diverso.
La conclusione non convince: se il principio attivo è diverso, EMA non avrebbe potuto e dovuto autorizzare il farmaco con la procedura semplificata richiesta per i biosimilari. Avrebbe invece dovuto richiedere i risultati delle sperimentazioni di fase I, di fase II e clinica ed autorizzarlo come originator (diverso) ancorché terapeuticamente equivalente.
L’errore, metodologico, sta a mio avviso nell’aver enfatizzato le differenze rimarcate da AIFA tra epoetina zeta ed epoetina alfa (originator): quelle differenze sono proprie di ogni biosimilare ed anzi di ogni farmaco biologico.
Anche il secondo position paper di AIFA, pure utilizzato nella motivazione della sentenza, vengono messe in rilievo le differenze. Però non si può sorvolare sul fatto che: a) tali differenze sono definite come “minori”, nel senso che non sono tali da spezzare il rapporto di equivalenza (forte) del biosimilare per degradarlo a rapporto di equivalenza (debole) tra due principi attivi diversi; b) sono dovute al peculiare metodo di produzione dei farmaci biologici.
Intervista a Dominique Van Doorne, vice Presidente di Accademia dei Pazienti
Come è nata l’Accademia dei pazienti e di che cosa si occupa?
In Italia l’Accademia dei pazienti onlus è stata creata allo scopo di promuovere il progetto europeo EUPATI (European Patients’ Academy on Therapeutic Innovation), a sua volta nato con l’obiettivo di insegnare al paziente tutto sulla ricerca e lo sviluppo dei farmaci, “dalla A alla Z”: dagli studi preclinici alle diverse fasi della ricerca clinica, fino alla fase IV quando il farmaco è già stato immesso in commercio.
Il progetto EUPATI è una partnership pubblico-privata fortemente voluta dalla Commissione europea, e in particolare dall’organismo comunitario denominato IMI (Innovative Medicines Initiative), in collaborazione con l’EFPIA (European Federation of Pharmaceutical Industries and Associations) e, soprattutto, con il Forum dei pazienti europei (EPF – European Patients’ Forum), che è stato uno dei soggetti promotori del progetto e ha assunto un ruolo di leadership. La considerazione di partenza che ha convogliato l’interesse di tutti è stata che non è possibile effettuare un numero sufficientemente alto di studi clinici in Europa se i pazienti, in primo luogo, non sono ben informati e consapevoli di che cosa sono gli studi clinici e che cosa comportano.
Il progetto EUPATI è stato inizialmente sviluppato in 12 paesi europei e tradotto in 7 lingue; adesso i paesi coinvolti sono 14 e i materiali sono disponibili in 9 lingue, ma la diffusione sta continuando a crescere.
Sono previsti tre livelli di formazione. Il primo livello ha come target il “paziente esperto” e si rivolge ad un numero ristretto di pazienti che ricevono una formazione molto intensa tramite un corso della durata di un anno, strutturato con 160 ore di formazione a distanza, in remoto, e una serie di incontri in presenza, face-to-face. Per ovvie ragioni logistiche (era molto complicato prevedere la partecipazione da Paesi diversi), nei corsi effettuati a livello europeo è stato possibile organizzare solo 2 incontri face-to-face, della durata di 4 giorni. Nel corso che attualmente si sta svolgendo in Italia e che vede la partecipazione di 54 pazienti, dalla Valle d’Aosta alla Sicilia, abbiamo deciso di organizzare 6 incontri face-to-face, un sabato al mese.
Il progetto EUPATI è attivo in 14 Paesi UE
Ai pazienti esperti, che costituiscono il primo livello del nostro progetto, spetta quindi il compito di formare, a loro volta, tutti i pazienti che afferiscono alle rispettive associazioni, promuovendo il diffondersi di una sempre maggiore cultura della ricerca e sviluppo dei farmaci, che permetta anche di interpretare e valutare correttamente, ad esempio, le informazioni che vengono riportate dai media, per comprendere la reale portata di alcuni articoli di giornale che annunciano nuove terapie disponibili e che, spesso, possono creare false illusioni o peggio ancora, se distorte, vere e proprie truffe. Il paziente formato, che conosce il metodo di ricerca e sviluppo dei farmaci, possiede una maggiore consapevolezza, ha gli strumenti per capire meglio quanto riportato dai giornali o dalla pubblicità e può riconoscere le situazioni non chiare.
Il terzo livello riguarda la promozione dei temi legati alla ricerca e sviluppo dei farmaci a tutti i cittadini, anche la popolazione sana in generale, con l’obiettivo di fornire le basi necessarie per capire, ad esempio, che cosa vuol dire “efficacia” o “sicurezza” di un farmaco e come vengono valutate; questo permette ai cittadini di discutere con il proprio medico con un livello di conoscenze e competenze più elevato.
A questo proposito è già disponibile per tutti, sul sito europeo di EUPATI e in traduzione italiana anche sul sito di Accademia dei pazienti, il cosiddetto “Toolbox” o “Cassetta degli attrezzi”, cioè una raccolta delle principali informazioni sulla ricerca e sviluppo dei farmaci che possono essere consultate online o scaricate e stampate in formato pdf.
Il nostro auspicio è che questo progetto possa avere un impatto positivo sull’aderenza terapeutica di tutti i pazienti: chiunque debba seguire una terapia deve essere consapevole del fatto che ogni farmaco funziona perché ha un meccanismo d’azione preciso e che la terapia deve essere assunta in un determinato modo perché gli errori nell’assunzione dei farmaci possono portare direttamente ad una inefficacia della terapia. Se prendiamo ad esempio le statine, è bene che i pazienti siano consapevoli del fatto che le statine non agiscono da un giorno all’altro, che la statina prescritta come prevenzione cardiovascolare non serve solo a ridurre il valore del colesterolo ma che la prevenzione funziona solo dopo un anno intero di terapia; per questo è importante fornire al paziente le informazioni necessarie a fargli comprendere che il suo obiettivo non deve essere solo la riduzione del valore di colesterolo a breve termine (“prendo le statine, faccio i prelievi, il valore è ridotto, sospendo le statine”) ma la prevenzione di eventi cardiovascolari a lungo termine: se il paziente lo sa, è motivato ad assumere correttamente la terapia.
In quest’ottica, un altro aspetto importante del nostro progetto è che la formazione non è legata ad una patologia specifica o ad un gruppo di pazienti in particolare, ma è trasversale perché non riguarda i farmaci o le terapie per una determinata malattia ma affronta il tema della metodologia di ricerca e sviluppo dei farmaci, che può applicarsi alle diverse aree terapeutiche.
Oltre al corso per pazienti esperti, Accademia organizza anche dei mini-corsi su misura, della durata di una giornata, per rispondere alla richiesta di una singola Associazione di pazienti o un Ospedale o un Centro di studi clinici in merito alla formazione su un argomento specifico (ad esempio, solo su HTA o solo su affari regolatori).
Quali risultati avete ottenuto a livello europeo e a livello italiano?
Il primo corso in inglese è stato reso disponibile nel 2015-2016: a livello europeo sono stati già formati 150 pazienti, e altri 50 pazienti sono in attivo. Tra i primi 150 pazienti formati a livello europeo, possiamo contare su 5 pazienti esperti italiani, che hanno frequentato il corso in inglese e stanno già fornendo la loro “consulenza” a diversi soggetti che ne hanno richiesto l’intervento, come ad esempio l’Agenzia europea del farmaco (EMA) oppure alcune aziende farmaceutiche, per il disegno di un protocollo clinico o la scrittura di un consenso informato. Alcuni sono anche attivi nei Comitati Etici o nelle strutture ospedaliere.
Come dicevo in precedenza, attualmente è attivo il primo corso in italiano e abbiamo 54 pazienti in formazione.
Per la filiera del farmaco le agenzie regolatorie, in Europa e in Italia, svolgono un ruolo fondamentale: sono state coinvolte nel vostro progetto?
Il coinvolgimento delle agenzie regolatorie nel nostro progetto è fondamentale, ed è stato duplice. Innanzitutto, nella fase di partenza, a livello europeo, le Agenzie regolatorie europee, così come tutti gli altri stakeholders interessati (aziende farmaceutiche, accademia e pazienti), sono state chiamate a verificare la correttezza, la trasparenza e la neutralità dei contenuti del corso.
Successivamente, per l’avvio del progetto anche a livello italiano, Accademia dei pazienti ha ritenuto importantissimo firmare un protocollo di intesa con AIFA. Già nel 2014 AIFA ha compreso la bontà del progetto che, formando pazienti affetti da diverse patologie, può fornire interlocutori autorevoli per AIFA: pertanto, il Direttore generale dell’epoca, Luca Pani, ha accettato immediatamente di stipulare un protocollo d’intesa con noi, che è stato confermato dal professor Melazzini e attualmente dal professor Li Bassi. Tanto è vero che AIFA ci invia dei relatori dell’Agenzia per il modulo relativo agli affari regolatori.
Gli aspetti legati all’economia del farmaco e alla sostenibilità del Servizio sanitario nazionale sono molto presenti nel dibattito pubblico: nel vostro corso i pazienti ricevono una formazione anche su questi temi?
Il nostro corso prevede un modulo specifico sui principi e l’applicazione dell’HTA (Health Technology Assessment), realizzato tramite un protocollo di intesa con l’ISPOR (International Society for Pharmacoeconomics and Outcomes Research), nella persona del professor Francesco Saverio Mennini.
Inoltre sul tema della farmacoeconomia abbiamo recentemente organizzato un webinar interamente dedicato al farmaco generico e al biosimilare per spiegare quali sono attualmente le regole in Europa e in Italia, quali sono le garanzie per il paziente e quali opportunità possono rappresentare questi farmaci in termini di risparmio per la sanità pubblica, anche nell’ottica di poter successivamente dedicare il risparmio così generato alla spesa per le innovazioni tecnologiche che, in questo momento, stanno arrivando in grande quantità.
A suo parere, quale può essere il ruolo del paziente esperto nel post-marketing?
Sicuramente i pazienti possono avere un ruolo attivo anche nella fase del post-marketing di un farmaco, soprattutto in relazione al tema della farmacovigilanza, che viene affrontato in un modulo del nostro corso. I nostri pazienti esperti capiscono in pieno qual è l’importanza della farmacovigilanza, in particolare nei primi 5 anni dall’immissione al commercio di un farmaco, sono in grado di valutare la numerosità del campione di pazienti sul quale vengono condotti gli studi clinici e conoscono le probabilità di insorgenza di diversi effetti collaterali, registrati o meno nel corso dei trial. In questo senso, sensibilizzati sulla farmacovigilanza, sanno che qualunque cittadino può segnalare un effetto collaterale, oltre che ai propri medici e al farmacista, anche direttamente all’AIFA, e questo è un aspetto che solitamente nessun cittadino conosce. Promuovere queste informazioni, capire che cos’è un effetto collaterale e sapere come compilare il modulo di segnalazione predisposto dall’AIFA riveste un forte impatto sul paziente, sul cittadino e sulla società intera.
Può esserci un ruolo per il paziente esperto anche nel procurement e nell’accesso al mercato?
Un ulteriore aspetto interessante di questo corso è che i pazienti formano un network, si conoscono e si confrontano fra di loro, si informano su tutto quello che accade in Italia. Il nostro compito come Accademia è di dare loro gli strumenti per consentirgli di organizzare il lavoro di lobbying, di pressione positiva sul sistema. Diventando più autorevoli, acquistano anche la capacità di arrivare a presentare le proprie istanze a livelli elevati, ad esempio discutendo con un Assessore regionale sul Prontuario e intervenendo laddove necessario. Ci auguriamo che i pazienti esperti formati dalla nostra Accademia possano creare un movimento per rendere il panorama più omogeneo in Italia e ridurre le disparità che ci sono attualmente. Questo è un compito che spetta a loro e alle Associazioni di pazienti. Un ruolo che Accademia dei pazienti vorrebbe invece portare avanti è quello di trovare le regole di ingaggio dei pazienti, ad esempio nei Comitati Etici. La nostra proposta è di istituire dei tavoli di discussione con le Istituzioni per capire quale possa essere il ruolo del paziente esperto e quali le modalità per un suo corretto ed efficace coinvolgimento. Purtroppo, al momento attuale, il paziente che entra nei Comitati Etici molto spesso riveste un ruolo passivo, è una figura che non è in grado di studiare il dossier di un farmaco, né di leggere un protocollo di studio clinico, né di capirne l’impatto o di comprendere i dati di tossicologia e della fase preclinica. Il paziente esperto che viene formato dal nostro corso è invece in grado di partecipare in maniera consapevole e utile, pertanto ci sembra opportuno che anche le Istituzioni prendano atto di potersi avvalere di questa nuova figura e stabiliscano che, per entrare a far parte di un Comitato Etico, anche il paziente debba attestare la sua competenza e formazione.
Nel vostro percorso, avete trovato qualche difficoltà in particolare?
Le difficoltà maggiori ci sono state soprattutto all’inizio del nostro progetto, perché abbiamo cominciato a lavorare sulla sensibilizzazione al tema della formazione dedicata al paziente e sulla promozione del corso nei diversi Paesi europei già dal 2012, quando ancora il materiale del corso non esisteva ed era in fase di preparazione (la prima versione è stata consegnata a partire dal 2015-2016). È stato quindi molto difficile e impegnativo promuovere il corso, organizzare il necessario fundraising, far capire di che cosa si trattava e preparare l’ambiente nei singoli Paesi affinché il corso, una volta pronto, fosse accolto in maniera favorevole. Questa è stata una scelta decisa dai promotori a livello europeo e possiamo ora dire che ha dato buoni frutti, ma all’epoca è stato molto faticoso.
E come sta andando la formazione per paziente esperto attualmente in corso in Italia?
Oggi il riscontro alla prima edizione del corso in italiano è veramente positivo e molti attori del sistema si stanno rivolgendo a noi e ci chiedono di partecipare. Ad esempio, sono interessate le Società scientifiche e molti medici ci chiamano per esprimere il loro apprezzamento all’iniziativa e chiedere di partecipare come uditori, per vedere personalmente che cosa succede durante gli incontri face-to-face. I relatori stessi del corso, che sono tutti esperti riconosciuti del settore (molti sono farmacologi), sono affascinati da questi pazienti che, essendo anche i primi, sono estremamente motivati e hanno già una preparazione di base piuttosto alta, che hanno dovuto acquisire da autodidatti, negli anni passati, in mancanza di una formazione specifica. Anche per me, che sono il direttore scientifico del corso, è un piacere veder intervenire questi pazienti confrontandosi con dei relatori di livello altissimo.
Sono inoltre lieta di poter riscontrare l’interesse della vostra rivista, che si rivolge anche alle farmacie ospedaliere e agli assessorati regionali, perché penso che gli Assessori e le farmacie potrebbero consultare i pazienti esperti nei processi decisionali e ricevere da loro un contributo positivo per tutto il sistema: le decisioni prese con la partecipazione attiva del paziente esperto potrebbero avere un impatto positivo sulla popolazione generale, poter affermare che nel tavolo di lavoro era presente anche un paziente esperto dà una garanzia e un valore aggiunto a tutti i pazienti, che si sentono maggiormente rappresentati e coinvolti nelle decisioni.
In Italia il primo corso per Paziente Esperto EUPATI è stato inaugurato il 24 novembre 2018 e terminerà a settembre 2019.
Modulo
Titolo
1
Scoperta e pianificazione dello sviluppo dei farmaci
2
Studi preclinici e sviluppo farmacologico
3
Sviluppo clinico esploratorio e confermatorio
4
Studi clinici
5
Affari regolatori, sicurezza dei medicinali, farmacovigilanza e farmacoepidemiologia
6
Principi e pratica dell’Health Technology Assessment (HTA)
Nel contesto attuale, la governance farmaceutica e, più in generale, quella del Servizio Sanitario Nazionale vengono influenzate in maniera consistente dal processo degli acquisti, nel quale intervengono numerosi fattori, con diverse finalità ed esigenze: innanzitutto, le normative sugli acquisti (il Codice degli appalti e i relativi correttivi – D.Lgs. N. 50/2016, D.Lgs 56/2017, D.L. 32/2019 – e le Circolari ANAC), quelle dedicate al settore farmaceutico su tetti e tipologia di assistenza (Legge finanziaria 2019 – L. 145/2018 ), ma anche le Sentenze dei Tribunali amministrativi e le Regioni, con i vari provvedimenti di indirizzo sulle politiche farmaceutiche. Inoltre, a livello regionale sono attive la Commissione per il Prontuario Terapeutico Regionale (farmaci e dispositivi medici), la Commissione di Health Technology Assessment, la Commissione protesica), oltre alle Aziende sanitarie (USL e ospedaliere) e alle eventuali aggregazioni tra aziende sanitarie.
Il percorso degli acquisti si sviluppa in diverse fasi, di competenza del farmacista (come Direttore esecuzione del contratto) e/o del provveditore (come Responsabile unico del provvedimento – RUP), e anche congiuntamente: la determinazione dei fabbisogni qualitativi e quantitativi, la conoscenza tecnico-merceologica dei prodotti, l’applicazione del Codice degli appalti e la scelta della procedura d’acquisto, la costruzione del capitolato di gara, la scelta del criterio di aggiudicazione (offerta economicamente più vantaggiosa o prezzo più basso), la determinazione dei requisiti di qualità e la relativa parametrazione, la Commissione di gara, la gestione del contratto e la valutazione degli esiti della gara.
Al centro del sistema si pone il paziente, nel cui interesse devono muoversi gli altri attori, al fine di garantire l’appropriatezza prescrittiva verso il singolo paziente e la scelta del farmaco più appropriato per la patologia specifica, l’approvvigionamento tempestivo, la libertà prescrittiva del medico, la continuità terapeutica e l’accesso all’innovazione.
Sono inoltre fondamentali alcuni elementi legati al contesto specifico locale quali il coinvolgimento dei medici specialisti prescrittori per la condivisione dei provvedimenti regionali, anche tramite accordi su specifici indicatori di spesa e di appropriatezza e la partecipazione alla costruzione del capitolato di gara e dei lotti, nonché la presenza di sistemi informatici per il monitoraggio delle prescrizioni e di sistemi di informazione continua dei medici specialisti prescrittori sui prezzi di acquisto/costi terapia.
Il Codice degli appalti ha innescato una rimodulazione del processo degli acquisti
Infine, nelle diverse esperienze di aggiudicazioni delle varie centrali di committenza si stanno evidenziando alcune criticità che vanno tenute presenti, come ad esempio le differenti condizioni di aggiudicazione (durata di validità e riapertura del confronto ad ogni uscita di un nuovo biosimilare, ecc..), i diversi prezzi ottenuti sul territorio nazionale, la disponibilità di diverse tipologie di farmaci (un unico farmaco o più farmaci), l’aggiudicazione del prodotto soltanto per pazienti naïve o per tutti, il ricorso soltanto al primo in graduatoria di un accordo quadro.
Con l’introduzione del Codice degli appalti, il processo degli acquisti è stato al centro di una rimodulazione da parte di tutti gli operatori.
La Società Italiana di Farmacia Ospedaliera e dei Servizi Farmaceutici delle Aziende Sanitarie (SIFO) e la Federazione delle Associazioni Regionali Economi e Provveditori della Sanità (FARE) stanno sviluppando un progetto dal titolo “Acquisto dei farmaci e dei dispositivi medici alla luce del Nuovo Codice degli appalti” con l’obiettivo di pervenire a regole condivise sulla strutturazione dei contenuti sostanziali del ciclo dell’appalto, dalla definizione dei fabbisogni all’esecuzione dei contratti e alla verifica degli esiti delle gare (mediante un confronto tra le diverse esperienze).
In quest’ottica vorremmo qui presentare alcune riflessioni sulla costruzione del capitolato di gara, soffermandoci su alcuni elementi della normativa e, in particolare, sui criteri di aggiudicazione e sulla valorizzazione della qualità.
Il primo punto da affrontare rimane la fase di determinazione dei fabbisogni, che ha assunto un nuovo e diverso significato per una serie di motivi: infatti, la normativa impone il rispetto dell’importo messo a gara e associato al contratto e al CIG (Codice Identificativo Gara); inoltre, nella gestione di farmaci ad alto costo, con possibilità di una grande variabilità della spesa, è necessario poter disporre di una previsione precisa della spesa da sostenere per il bilancio aziendale; infine, definire con precisione un fabbisogno qualitativo e quantitativo consente di determinare i contratti per tutti i prodotti da utilizzare, garantendone una tempestiva messa a disposizione. La definizione dei fabbisogni “qualitativi” viene effettuata tramite la Commissione terapeutica, mentre la definizione dei fabbisogni “quantitativi” si poggia sull’analisi dello storico, su analisi previsionali e su modelli statistici. La normativa attuale assegna alla Commissione terapeutica un nuovo compito, cioè determinare le conseguenze correlate con l’inserimento di nuovi farmaci e/o dispositivi medici (DM) sia in termini economici sia in termini di utilizzo: quanti e quali farmaci e/o DM entreranno nel Prontuario, quanti e quali saranno sostituiti? Si impone quindi la necessità di integrare l’attività della Commissione del Prontuario Terapeutico Regionale con quella di chi è preposto all’elaborazione di un capitolato di gara e con quella svolta dalla Regione. In quest’ottica, risulterebbe opportuno da parte degli operatori avvalersi del contributo delle ditte produttrici che hanno a disposizione dati indispensabili per determinare le suddette valutazioni sui nuovi farmaci e DM ma che sono di difficile reperibilità, come dati epidemiologici e analisi di budget impact.
Un altro punto sul quale soffermarsi è l’equivalenza terapeutica: a questo proposito, il progetto SIFO-FARE suggerisce di stabilire un percorso univoco che, attraverso la corretta interpretazione della normativa e le sentenze di tribunali amministrativi e del Consiglio di Stato, arrivi ad elaborare un algoritmo per ogni gruppo di farmaci su come poter associare il più appropriato capitolato di gara, in un contesto costantemente aggiornato sulla base delle più recenti evidenze scientifiche.
L’art. 51 del Codice degli appalti definisce la suddivisione in lotti e ne affida il compito alle stazioni appaltanti. Sotto il profilo concorrenziale, nel formare i lotti vanno tenuti presenti i seguenti criteri di ordine generale (Antitrust – segnalazione 28/9/1999):
individuare il giusto numero di lotti;
individuare la giusta dimensione di lotti: non ricomprendere nell’oggetto della gara più attività che, prese singolarmente, esplicano una funzione economica o tecnica e che quindi potrebbero da sole costituire oggetto di appalto; non frazionare il progetto in singoli lotti al fine di eludere la disciplina comunitaria sugli appalti pubblici se ciò comporta che alcuni di essi siano poi sprovvisti di una propria funzione economica o tecnica;
favorire la partecipazione di nuovi concorrenti: il relativo valore deve essere adeguato in modo da garantire l’effettiva possibilità di partecipazione da parte di microimprese, piccole e medie imprese;
aggiudicare il lotto a due (o più) fornitori, che resteranno in competizione per l’intera durata contrattuale;
aggregare la domanda tramite accorpamenti di fabbisogni;
valutare attentamente la formazione di lotti composti (stesso principio attivo e più formulazioni/dosaggi);
motivare obbligatoriamente la mancata suddivisione in lotti.
La determinazione del prezzo a base d’asta risulta essere un passaggio fondamentale perché, da un lato, deve garantire la partecipazione a più concorrenti possibile e, dall’altro, deve garantire di ottenere un prezzo di aggiudicazione più basso possibile. Pertanto, tale equilibrio non può essere lo stesso per tutti i tipi di prodotti e il prezzo a base d’asta va determinato di volta in volta a seconda della tipologia di farmaci da mettere in concorrenza. Occorre conoscere il mercato, il numero dei concorrenti, i risultati delle gare di altre stazioni appaltanti, la significatività dell’importo messo a gara rispetto a quello delle altre stazioni appaltanti e poi dedurre la soluzione più appropriata. La normativa ci dice che il prezzo base d’asta dovrà essere “il prezzo di mercato”, vale a dire, il prezzo di aggiudicazione delle diverse centrali di committenza, il più basso o il prezzo medio oppure, per le molecole per le quali esiste il biosimilare, il prezzo più basso di registrazione dei biosimilari.
Vale la pena riportare come esempio quello dei farmaci oncologici genericabili endovenosi, laddove si assiste a tutt’oggi a gare con un prezzo base d’asta sempre più basso, in una situazione in cui i prezzi di registrazione dei farmaci sono già molto bassi e i risultati delle gare sono lotti deserti o interruzione della fornitura.
Nel capitolato di gara può essere prevista la possibilità di accettare offerte che superino la base d’asta in maniera tale di garantirsi la possibilità di ricorrere ad alcune molecole per eventuali esigenze (continuità terapeutica e/o nel caso in cui non ci siano offerte al di sotto della base d’asta prevista).
Focalizzandoci sulla valorizzazione della qualità, il Codice degli appalti ha introdotto il criterio dell’offerta economicamente più vantaggiosa (OEPV) sulla base del miglior rapporto qualità/prezzo che consente:
di assegnare un’importanza preminente al profilo tecnico-qualitativo rispetto a quello economico;
di non aggiudicare l’appalto ad un’offerta che, ancorché conveniente sotto il profilo economico, non sia apprezzabile sotto il profilo tecnico;
di fissare una soglia di sbarramento per l’aspetto tecnico- qualitativo al di sotto della quale le offerte non saranno valutate;
di inserire la qualità dell’offerente tra i criteri di aggiudicazione.
Pertanto, diventa cruciale la definizione di che cosa valutare ed è necessario fissare un livello qualitativo minimo e un livello di qualità massima valorizzabile, considerando che bisogna evitare di pagare un prezzo alto per una qualità ridondante rispetto all’uso previsto: si deve valorizzare non la qualità assoluta, ma la qualità utile nel contesto dato.
Ai due estremi, rimane possibile l’aggiudicazione facendo prevalere il criterio del minor prezzo, o valorizzando solo parametri qualitativi.
Il criterio di miglior rapporto qualità/prezzo costituisce per le imprese, nel lungo-medio periodo, un’opportunità per far emergere le loro capacità, assicurando una maggiore concorrenza, ma nell’immediato richiede maggiori oneri per la formulazione delle offerte tecniche.
Redditività, costo di utilizzazione e costo dell’intero ciclo di vita del prodotto diventano parametri valutativi imprescindibili per la determinazione del costo totale di un prodotto. Si impone dunque di identificare un differenziale tra prodotti diversi che abbiano (o meno) principi attivi diversi e, se questo differenziale esiste, andrà correttamente identificato, valutato, qualificato, misurato, pesato e inserito in un sistema di ponderazione.
Il concetto di qualità viene a dir poco “esploso” e dovrà essere declinato nei suoi vari aspetti (Figura 1):
qualità;
organizzazione, qualifiche ed esperienza;
servizi post-vendita e assistenza tecnica.
Figura 1. Possibile evoluzione del concetto di qualità con il Codice degli appalti
Nel contesto del Progetto SIFO-FARE, con il contributo dei membri del Board e del tavolo tecnico “Farmaci”, è stata elaborata una “Scheda di qualità” che riporta i parametri qualitativi relativi ai farmaci, alle apparecchiature e/o ai DM e ai servizi, individuando una serie di piani e competenze (Tabella I e Appendice) che hanno l’obiettivo di supportare gli operatori nella valorizzazione della qualità in fase di costruzione del capitolato.
Parametri qualitativi legati al farmaco
Piano farmaceutico
Commissione di gara
Piano farmacologico
Piano tossicologico
Piano regolatorio
Commissione terapeutica
Parametri qualitativi legati ad apparecchiature e/o dispositivi medici
Piano tecnologico
Commissione di gara
Parametri qualitativi legati ai servizi correlati
Piano logistico
Piano di assistenza post-vendita
Commissione di gara
Tabella I. Struttura della Scheda qualità elaborata dal progetto SIFO-FARE
(La scheda di qualità per intero si riporta in Appendice)
Il cosiddetto Correttivo appalti, cioè il D.Lgs. 56/2017, ha introdotto un tetto massimo per il punteggio economico entro il 30%, con la conseguenza che il punteggio attribuito alla qualità e ai suoi requisiti risultava almeno pari al 70%.
Nell’ambito del progetto SIFO-FARE si è predisposto un documento a firma congiunta con Assobiomedica, Assogenerici, Cittadinanzattiva e Farmindustria, in cui sono state riportate alcune criticità sul Codice degli appalti con relative proposte correttive. In particolare, tra le altre proposte emendative, il documento conteneva una richiesta di modifica del comma 10 bis, art. 95 scaturita dalla considerazione che, a seguito del tetto massimo del 30% per il punteggio economico introdotto dal Correttivo appalti, nella pratica si è verificato un maggior utilizzo del criterio del minor prezzo a discapito della valorizzazione della qualità, con un effetto contrario all’obiettivo di promozione della qualità previsto dal legislatore.
Nel documento SIFO-FARE si auspicava dunque che il Codice fornisse indicazioni di utilizzo per macro tipologie di prodotti, in particolare nella fornitura di beni e servizi in ambito sanitario, con applicazione di rapporti prezzo/qualità diversificati, rimessi alla discrezionalità delle stazioni appaltanti. Tale documento è stato consegnato nei primi giorni di aprile al Capo di Gabinetto del Ministero della Salute.
L’obiettivo è valorizzare non la qualità assoluta ma la qualità utile nel contesto dato
Pochi giorni dopo si è avuta una prima modifica del Codice, con il Decreto Legge n. 32 del 18 aprile 2019, che ha previsto un cambio sostanziale di orientamento, accogliendo la modifica del tetto massimo del 30% per il punteggio economico e riconducendo la valutazione alle stazioni appaltanti. Inoltre il Decreto Legge n. 32 ha ripristinato il ricorso al prezzo più basso come prima opzione per il criterio di aggiudicazione, stabilendo che il ricorso all’offerta economicamente più vantaggiosa debba avvenire «previa motivazione»; nella precedente normativa, era il prezzo più basso a dover essere motivato dalla stazione appaltante che intendeva avvalersene. Per non andare contro la direttiva comunitaria ed evitare eventuali infrazioni, la modifica si applica solo agli appalti sotto soglia comunitaria.
Appendice: scheda qualità elaborata dal progetto SIFO-FARE con il contributo dei membri del Board e del tavolo tecnico “Farmaci”
Parametri qualitativi legati al farmaco
Piano farmaceutico *
Forme farmaceutiche che favoriscono compliance del paziente etc.